
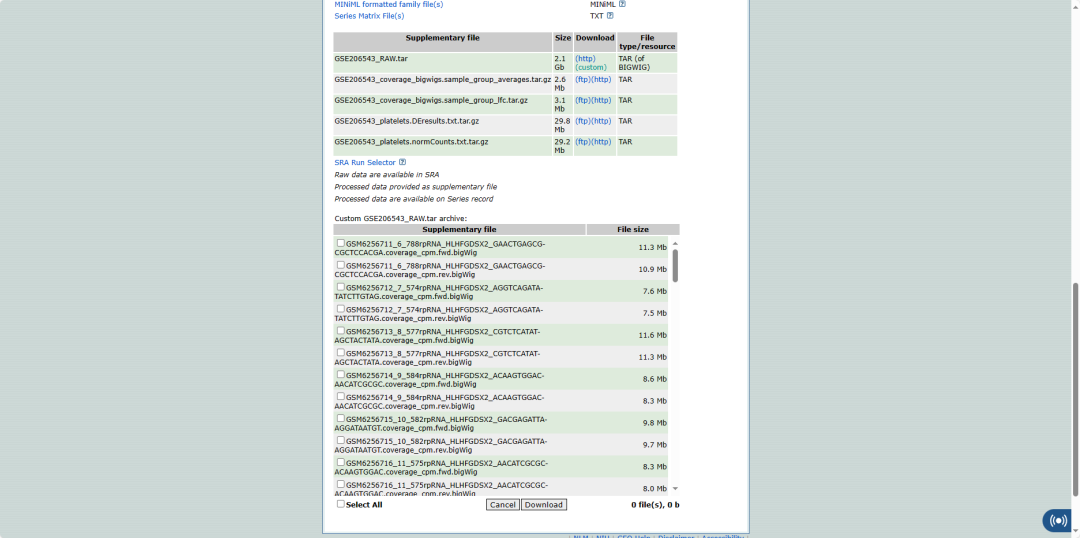

看到一篇文章的测序数据主要提供了bigwig文件,然后就试着处理了一下。
BigWig文件
BigWig文件是一种用于存储大规模测序数据(如RNA-seq、ChIP-seq等)的广泛使用的文件格式。它是BigBed文件的一个变体,专门用于存储测序读取的覆盖信息,即每个基因组位置的读取计数。BigWig文件可以提供与Wiggle格式相同的信息,但是以更紧凑和高效的二进制格式存储,使得它们适合于存储和传输大型数据集。
推荐阅读:
- deepTools: tools for exploring deep sequencing data
https://deeptools.readthedocs.io/en/latest/#原文里面用的就是这个(deepTools 3.5.0)。 - Pohl, Andy, and Miguel Beato. “bwtool: a tool for bigWig files.” Bioinformatics (Oxford, England) vol. 30,11 (2014): 1618-9. doi:10.1093/bioinformatics/btu056
- Wilks, Christopher et al. “Megadepth: efficient coverage quantification for BigWigs and BAMs.” Bioinformatics (Oxford, England) vol. 37,18 (2021): 3014-3016. doi:10.1093/bioinformatics/btab152
代码思路
BigWigs to count matrix:
https://lcolladotor.github.io/protocols/bigwig_DEanalysis/#参考代码参考基因组及其注释下载方法:
https://zhuanlan.zhihu.com/p/383397412#Ensemble下载GRCm39:http://asia.ensembl.org#下载基因注释文件gtf即可。代码里面读长固定为76(不确定对不对,待查)。 #平均读长(read length):测序的下机数据里,所有序列的平均长度。以碱基(bp)为单位,常见的读长有50 bp、90 bp、100bp、150 bp。
#在RNA-seq(RNA测序)分析中,”fwd”(正向)和”rev”(反向)通常指的是单端或配对端RNA测序数据的方向性。”fwd”数据可能用于评估基因表达水平,而”rev”数据可能用于研究转录本的剪接模式。
得到Matrix之后,顺便用DEseq2进行差异分析。 #差异分析教程:
https://www.reneshbedre.com/blog/deseq2.html
代码
#目的:将bigwig文件转换成matrix数据,并进行下游差异分析
# 参考:https://lcolladotor.github.io/protocols/bigwig_DEanalysis/
## Locate bigWig files
files <- list.files('GSE206543_RAW/')
head(files)
## Find exons
gtf <- rtracklayer::import('Mus_musculus.GRCm39.111.chr.gtf.gz')
exons <- gtf[gtf$type=='exon']
## Import data and create count matrix
library('rtracklayer')
library(tidyverse)
library(data.table)
bw <- BigWigFileList(files)
# 遍历每个BigWig文件
for(i in 1:length(bw)) { #代码写得有点烂,速度有点慢
# 导入BigWig文件
chr <- import(str_c('GSE206543_RAW/', bw[[i]]@resource), as = 'RleList')
counts <- matrix(NA, nrow = length(exons), ncol = length(chr))
colnames(counts) <- names(chr)
# 遍历每个外显子区域
for(j in seq_len(length(chr))) {
# 获取当前外显子的覆盖度
coverage <- chr[[j]]
# 将计算结果赋值给计数矩阵的相应位置
counts[, j] <- sum(Views(coverage, ranges(exons)))
}
## Divide by read length and round to integer numbers,这个76不确定对不对。
counts <- round(counts / 76, 0)
#在所有染色体中选取表达量最高的基因
max_count <- data.frame(sapply(1:nrow(counts), function(i) max(counts[i, ])))
#表达太少的基因直接删掉,提高速度
max_count <- subset(max_count,max_count[,1] >= 10)
#根据行名合并数据集
if (i==1) {
all_count <- max_count
all_count$rn <- row.names(max_count)
} else {
max_count$rn <- row.names(max_count)
all_count <- merge(all_count,max_count,by='rn',all=T)
}
}
# 处理NA值
all_count[is.na(all_count)] <- 0
gene_id <- data.frame(exons$gene_name)
gene_id$rn <- rownames(gene_id)
expr <- merge(all_count,gene_id,by='rn')
rownames(expr) <- expr$rn
expr <- expr[,2:9]
#修改列名
sp_names <- data.frame(sapply(files, function(file) str_sub(file, 1, 10)))
colnames(expr) <- c(sp_names[,1],'gene')
#利用aggregate函数,对相同的基因名按列取取最大值
expr_max=aggregate(.~gene,max,data=expr)
#把表达矩阵写出来
fwrite(expr_max,file = "expr_max.csv")
###### Perform DESeq2 analysis #####
#DESeq2差异分析教程:https://www.reneshbedre.com/blog/deseq2.html
library('DESeq2')
rownames(expr_max) <- expr_max$gene
expr_max <- as.matrix(expr_max[,2:8])
coldata <- data.frame(sample=sp_names[,1],
condition=c(rep('RP',3),rep('MK',4)),
row.names = "sample" )
fwrite(group,file = "metadata.csv")
coldata$condition <- as.factor(coldata$condition)
#计数矩阵中列的名称必须与样本名称中的顺序相同
all(rownames(coldata) %in% colnames(expr_max))
all(rownames(coldata) == colnames(expr_max))
## Round matrix and specify design
dds <- DESeqDataSetFromMatrix(expr_max,colData = coldata,
design = ~ condition)
# set control condition as reference,MK是对照组
dds$condition <- relevel(dds$condition, ref = "MK")
dds <- DESeq(dds)
# see all comparisons (here there is only one)
resultsNames(dds)
res <- results(dds)
view(res)
write.csv(as.data.frame(res[order(res$padj),] ), file="dge.csv")
over √.
声明:文中观点不代表本站立场。本文传送门:https://eyangzhen.com/414710.html
