
论文
StainedGlass: interactive visualization of massive tandem repeat structures with identity heatmaps
代码链接
这个工具是用来可视化展示基因组水平上tandem repeat 的相似性,是用snakemake搭建的一个流程,今天的推文我们试着拆解一下这个流程里都有哪些步骤
这个流程依赖的软件是通过搭配conda配置文件的方式去安装,但是在集群上的计算节点很多时候是不能联网的,所以最好还是提前配置好依赖软件,依赖的软件在 workflow/env目录下的env.yaml和R.yaml下
- pandas
- numpy
- numba
- cooler
- minimap2==2.18
- bedtools
- samtools>=1.9
- pysam
- snakemake>=7.8
- snakefmt
- bwa
- pigz - xorg-libx11
- xorg-libxau
- r-base>=4.0
- r-essentials
- r-cairo
- r::r-tidyverse
- r-data.table
- r-cowplot
- r-argparse>=2.1.2
- r-glue
- r::r-rcolorbrewer
- r::r-scales
- r::r-ggplot2
- r-r.utils把依赖的软件和R包都安装一下
运行命令
snakemake -s ~/biotools/StainedGlass/workflow/Snakefile --configfile=/home/myan/biotools/StainedGlass/config/config.yaml --config sample=A fasta=/data/myan/raw_data/practice/stainedGlass/chr8_cen.fasta --cores 8 make_figures -pn会展示出这个流程每一步具体执行的命令,然后我们分别执行其中的命令看看每一步具体做了什么事
首先是对输入数据进行索引
samtools faidx chr1.fabedtools利用fai文件生成bed文件
## -s 参数可以设置滑窗 -w设置的是步长
bedtools makewindows -g chr1.fa.fai -w 2000 > output.bedbedtools根据bed文件分隔fasta文件
bedtools getfasta -fi chr1.fa -bed output.bed > output.2000.fastabatch_bed_files.py 这个脚本的作用好像是把bed文件进行分隔,–outputs参数后好像可以自定义写多少个输出
python ../batch_bed_files.py output.bed --outputs a0.bed a1.bed a2.bed这一步是对参考构建数据库
minimap2 -f 1000 -s 400 -ax ava-ont -d output.fasta.mmi output.2000.fasta这里的-f和-s参数没看懂是什么意思
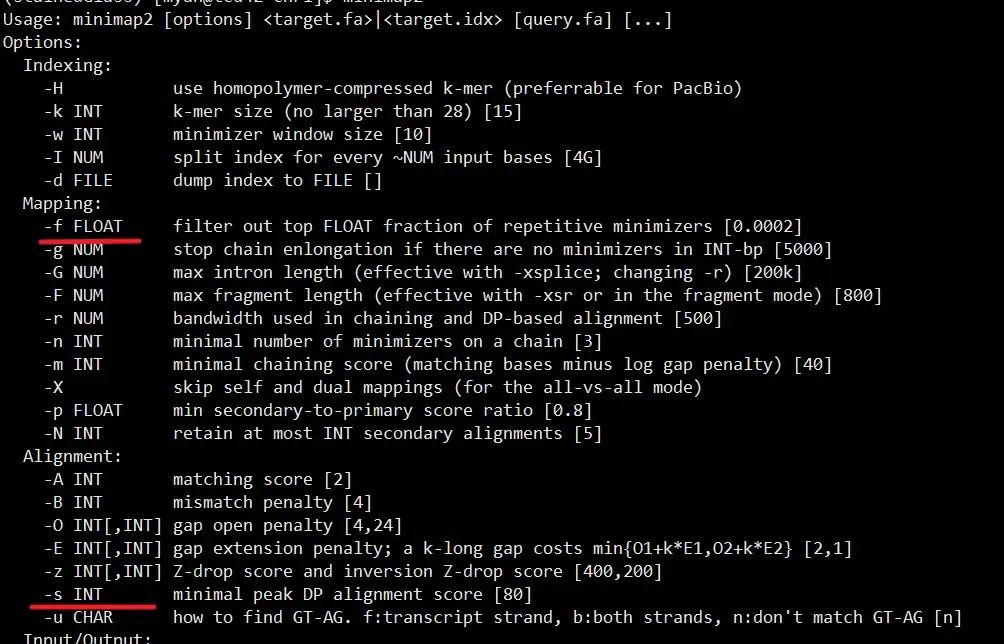
minimap2的帮助文档

根据分隔的bed文件分别提取fasta序列
bedtools getfasta -fi chr1.fa -bed a0.bed > a0.fa
bedtools getfasta -fi chr1.fa -bed a1.bed > a1.fa
bedtools getfasta -fi chr1.fa -bed a2.bed > a2.faminimap2比对生成bam文件并合并
minimap2 -t 4 -f 10000 -s 400 -ax ava-ont --dual=yes --eqx output.fasta.mmi a0.fa | samtools sort -m 4G -o a0.bam
minimap2 -t 4 -f 10000 -s 400 -ax ava-ont --dual=yes --eqx output.fasta.mmi a1.fa | samtools sort -m 4G -o a1.bam
minimap2 -t 4 -f 10000 -s 400 -ax ava-ont --dual=yes --eqx output.fasta.mmi a2.fa | samtools sort -m 4G -o a2.bam
samtools merge -@ 4 -O BAM merged.bam a0.bam a1.bam a2.bam
samtools index merged.bam接下来是画图,这里的两个python脚本起到了什么作用暂时还不太明白
python samIdentity.py --threads 8 --matches 400 --header merged.bam > output.tbl
bgzip -c output.tbl > output.tbl.gz
python refmt.py --window 2000 --fai chr1.fa.fai --full output.full.tbl.gz output.tbl.gz full.bed.gz
mkdir -p results/abc_figures/pdfs
mkdir -p results/abc_figures/pngs
Rscript aln_plot.R -b full.bed.gz --threads 8 --prefix abc输出的部分结果
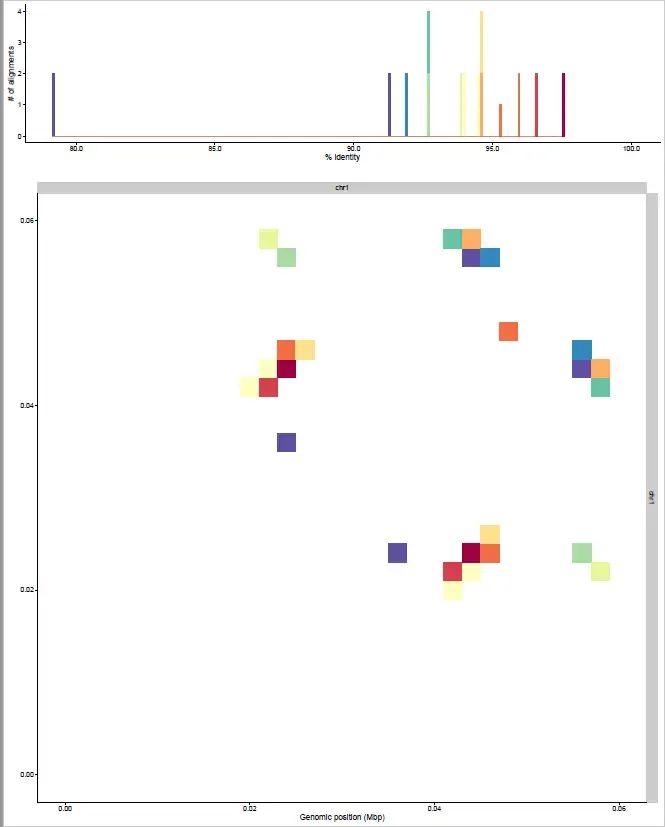

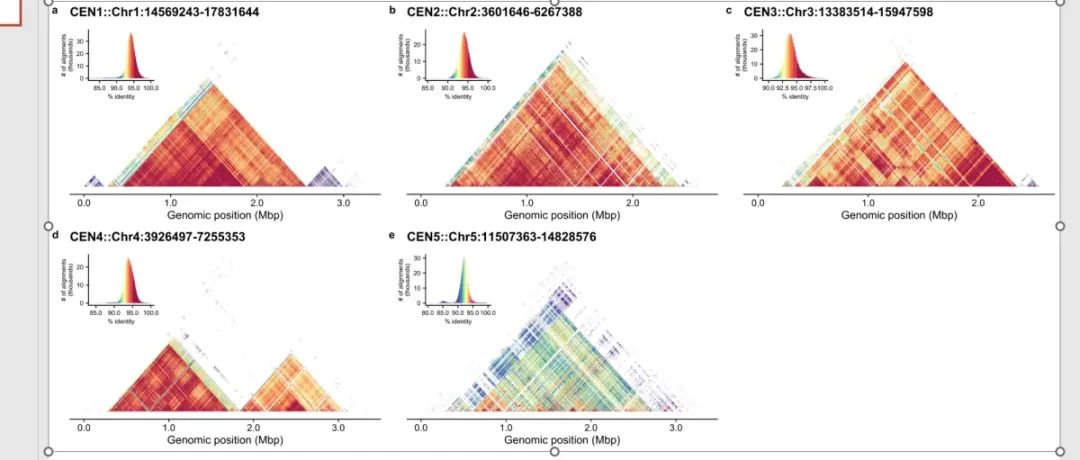
这个是论文中提供的图
推文记录的是自己的学习笔记,很可能存在错误,请大家批判着看
声明:文中观点不代表本站立场。本文传送门:https://eyangzhen.com/155771.html


