
论文
A graph-based genome and pan-genome variation of the model plant Setaria
谷子图基因组NG.pdf
代码
论文中的方法 Demographic history inference 部分 的代码也是可以找到的,今天的推文我们试着重复一下这部分的代码,代码的链接是
https://htmlpreview.github.io/?https://github.com/qiangh06/Setaria-pan-genome/blob/main/Population%20genomic%20and%20Demographic%20inference/Scripts.html
也可以从这个链接下载本地文件
vcf文件我们使用
这个链接中的 test.vcf.gz 文件 这个应该是番茄的数据,只有1号染色体,总共是 79982 个位点 516个样本
首先是应用plink对数据进行过滤
plink --vcf test.vcf.gz --set-missing-var-ids @:# --allow-extra-chr --biallelic-only strict --double-id --make-bed --out chr1
plink --bfile chr1 --allow-extra-chr --hardy --out chr1
awk '$7 < 0.05 {print $2}' chr1.hwe > het_filt_chr1.snps
plink --bfile chr1 --allow-extra-chr --extract het_filt_chr1.snps --make-bed --out chr1_het
plink --bfile chr1_het --allow-extra-chr --maf 0.05 --make-bed --out chr1_het_maf
plink --bfile chr1_het_maf --allow-extra-chr --geno 0.1 --mac 1 --make-bed --out chr1_het_maf_geno
plink --bfile chr1_het_maf_geno --allow-extra-chr --indep-pairwise 10kb 1 0.8 --out ld1_unadmixed_maf_1
plink --bfile chr1_het_maf_geno --allow-extra-chr --extract ld1_unadmixed_maf_1.prune.in --make-bed --out chr1_het_maf_geno_10kb
plink --bfile chr1_het_maf_geno_10kb --allow-extra-chr --indep-pairwise 50 1 0.8 --out ld2_unadmixed_maf_1
plink --bfile chr1_het_maf_geno_10kb --allow-extra-chr --extract ld2_unadmixed_maf_1.prune.in --make-bed --out chr1_het_maf_geno_10kb_50
然后用convertf这个命令做一个文件格式转换
convertf -p het_maf.parfilehet_maf.parfile 文件的内容
genotypename: chr1_het_maf_geno_10kb_50.bed
snpname: chr1_het_maf_geno_10kb_50.bim
indivname: chr1_het_maf_geno_10kb_50.fam
outputformat: PACKEDANCESTRYMAP
genooutfilename: tomato_het_maf.geno
snpoutfilename: tomato_het_maf.snp
indoutfilename: tomato_het_maf.ind前三行是输入文件,后三行是输出文件
convertf 这个命令在eigensoft这个工具里,可以直接使用conda安装
conda install eigensoft输出的ind文件最后一列是问号,需要替换成分组信息,就是那个个体是来源于哪个群体,我这里没有找到这个信息,就随便构造了
接下来的内容就是在R语言里操作了
admixtools这个R包的文档
各种统计量的简介 f2 f3 f4等
library(admixtools)
f2_blocks<-f2_from_geno("D:/Jupyter/practice/my_f2_dir/tomato_het_maf")
qpg_result<-qpgraph(f2_blocks,graph = matrix(c('R', 'R', 'n1', 'n1', 'n2', 'n2',
'pop4', 'n1', 'pop2', 'n2', 'pop3', 'pop1'), , 2),
return_fstats = TRUE)
qpg_result
qpg_result$score
qpg_result$f3
plot_graph(qpg_result$edges)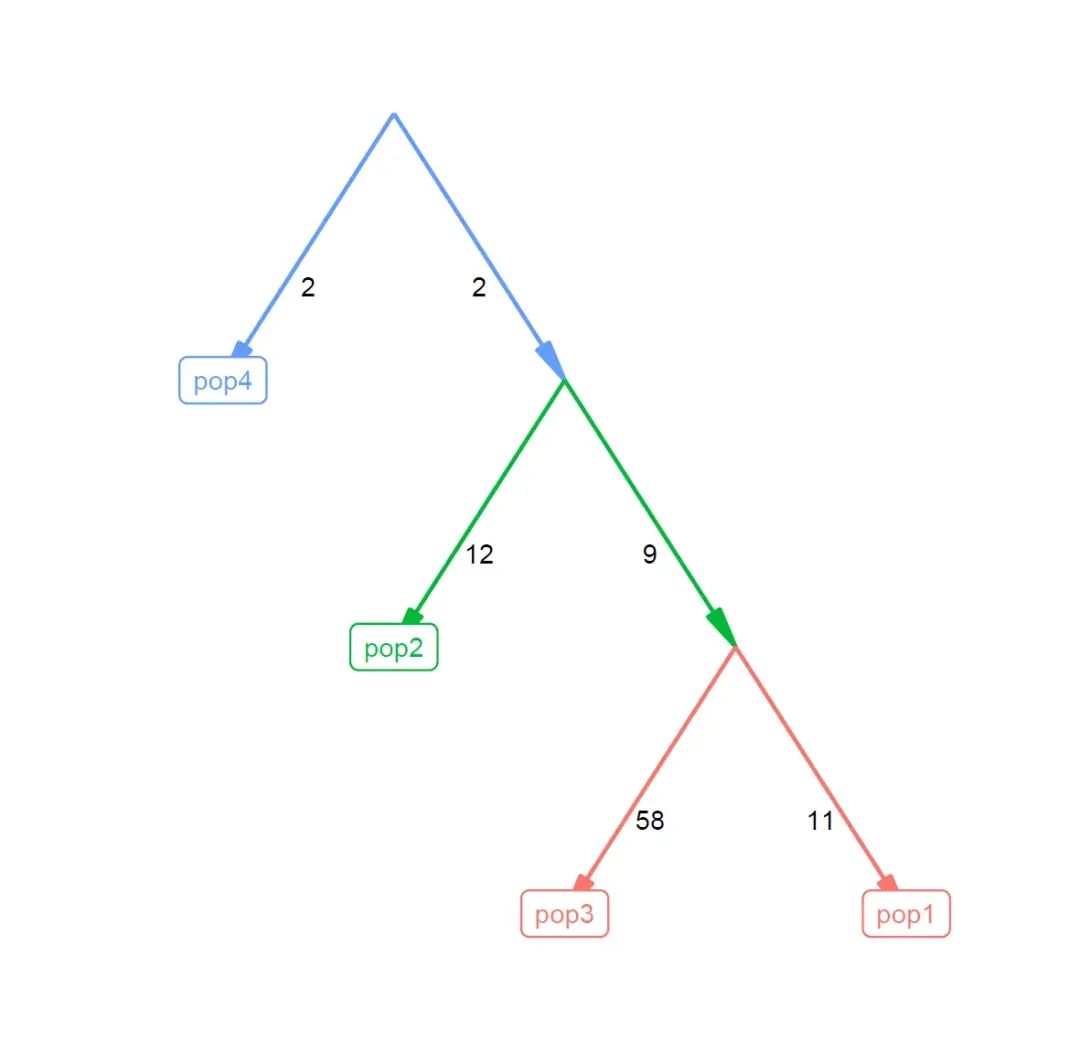

这部分论文中提供的代码应该是少了几个步骤
能跑通这个流程,但是很多内容都不理解,而且最终的结果也不会看,还得仔细看帮助文档,仔细看论文

推文记录的是自己的学习笔记,大概率存在错误,欢迎大家批评指正
声明:文中观点不代表本站立场。本文传送门:https://eyangzhen.com/225873.html


