
1引言
想着 ClusterGVis 在给热图添加注释的图形好像有些限制, 要是能自定义往每个 cluster 添加相关的图形就好了,不局限于热图里面的数据。灵感来自于推文 当 ComplexHeatmap 遇上 ggplot2 能碰出什么样的火花?。
所以你只需要画出图形(ggplot2),然后插入到相应的 cluster 里面就好了,我的第一个创意是: 比如单细胞展示 marker 基因时,在右边插入 marker 基因的散点图(featureplot) 。当然其它任何图形都可以了。
然后就是给 ClusterGVis 加了一个 gglist 的参数,你给一个 ggplot 的 list 就行了,注意顺序得对应好。
2安装
重新安装获取新功能:
# Note: please update your ComplexHeatmap to the latest version!
# install.packages("devtools")
devtools::install_github("junjunlab/ClusterGVis")3示例
准备一下数据:
library(ClusterGVis)
library(scRNAtoolVis)
library(org.Hs.eg.db)
library(ggplot2)
load("pbmc.rda")
# add cell type
new.cluster.ids <- c("Naive CD4 T", "CD14+ Mono", "Memory CD4 T", "B", "CD8 T", "FCGR3A+ Mono",
"NK", "DC", "Platelet")
names(new.cluster.ids) <- levels(pbmc)
pbmc <- RenameIdents(pbmc, new.cluster.ids)
# find markers for every cluster compared to all remaining cells
# report only the positive ones
pbmc.markers.all <- Seurat::FindAllMarkers(pbmc,
only.pos = TRUE,
min.pct = 0.25,
logfc.threshold = 0.25)
# get top 10 genes
pbmc.markers <- pbmc.markers.all %>%
dplyr::group_by(cluster) %>%
dplyr::top_n(n = 4, wt = avg_log2FC)
# prepare data from seurat object
st.data <- prepareDataFromscRNA(object = pbmc,
diffData = pbmc.markers,
showAverage = TRUE)画个简单的热图:
# heatmap plot
pdf('sc_g.pdf',height = 10,width = 10,onefile = F)
visCluster(object = st.data,
plot.type = "both",
line.side = "left",
column_names_rot = 45,
markGenes = pbmc.markers$gene,
cluster.order = c(1:9))
dev.off()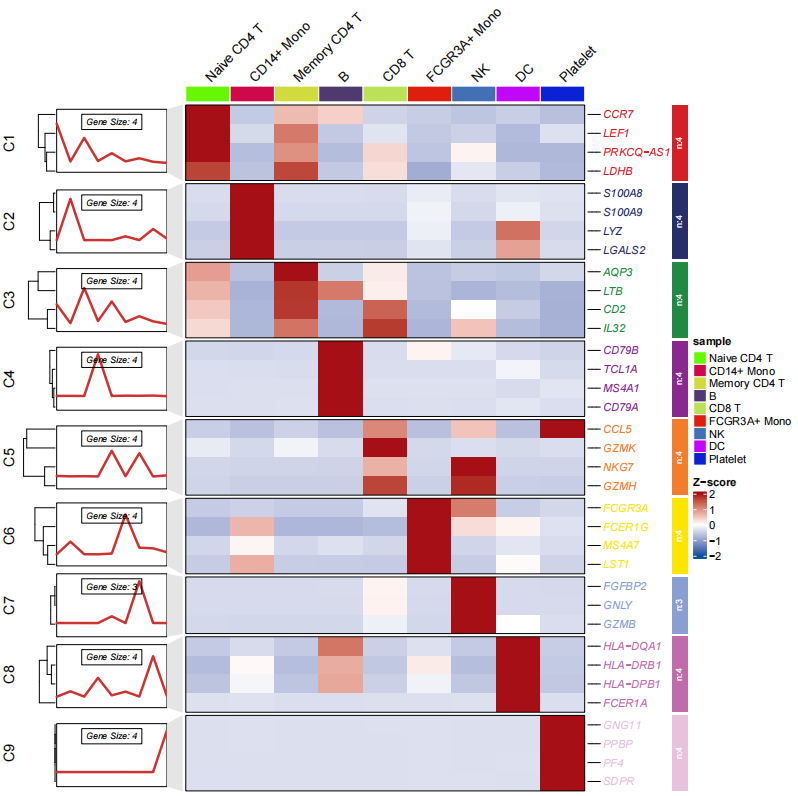

然后我们可以画这些基因的散点图:
# loop plot
lapply(unique(pbmc.markers$cluster), function(x){
tmp <- pbmc.markers |> dplyr::filter(cluster == x)
# plot
p <- Seurat::FeaturePlot(object = pbmc,
features = tmp$gene,
ncol = 4)
return(p)
}) -> gglist
# assign names
names(gglist) <- paste("C",1:9,sep = "")
然后就可以插入到图里了,这样右边就是对应 marker 基因在不同亚群的表达情况了:
# insert scatter plot
pdf('sc_ggplot.pdf',height = 16,width = 18,onefile = F)
visCluster(object = st.data,
plot.type = "both",
line.side = "left",
column_names_rot = 45,
markGenes = pbmc.markers$gene,
cluster.order = c(1:9),
ggplot.panel.arg = c(4,1,24,"grey90",NA),
gglist = gglist)
dev.off()
当然你也可以用 scRNAtoolVis 来画散点图:
# loop plot
lapply(unique(pbmc.markers$cluster), function(x){
tmp <- pbmc.markers |> dplyr::filter(cluster == x)
# plot
p <-
FeatureCornerAxes(object = pbmc,
reduction = 'umap',
groupFacet = NULL,
relLength = 0.65,
relDist = 0.2,
cornerTextSize = 2.5,
features = tmp$gene,
show.legend = F)
return(p)
}) -> gglist
# assign names
names(gglist) <- paste("C",1:9,sep = "")
# insert scatter plot
pdf('sc_ggplot_scRNAtoolVis.pdf',height = 16,width = 16,onefile = F)
visCluster(object = st.data,
plot.type = "both",
line.side = "left",
column_names_rot = 45,
markGenes = pbmc.markers$gene,
cluster.order = c(1:9),
ggplot.panel.arg = c(4,1,12,"grey90",NA),
gglist = gglist)
dev.off()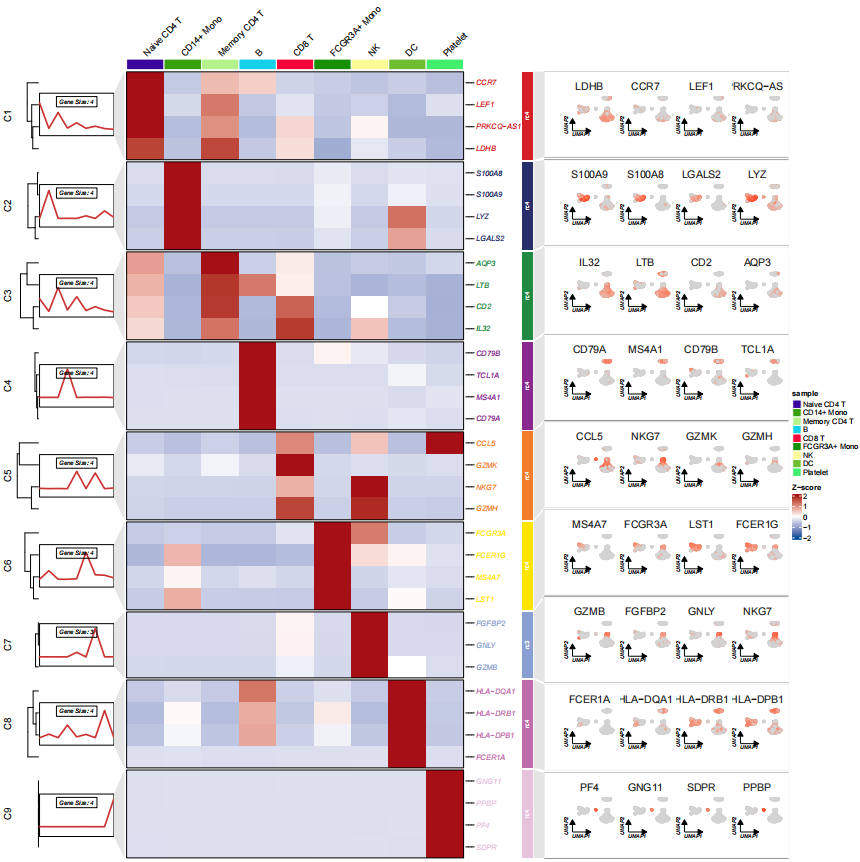
当然你也可以添加自定义的富集图,富集,当然这里只是演示,这四个基因富集也不能说明什么:
# enrich for clusters
enrich <- enrichCluster(object = st.data,
OrgDb = org.Hs.eg.db,
type = "BP",
organism = "hsa",
pvalueCutoff = 0.5,
topn = 5,
seed = 5201314)
# check
head(enrich,3)
# group Description pvalue ratio
# GO:0002573 C1 myeloid leukocyte differentiation 0.0003941646 66.66667
# GO:0050870 C1 positive regulation of T cell activation 0.0005222343 66.66667
# GO:1903039 C1 positive regulation of leukocyte cell-cell adhesion 0.0006265618 66.66667绘制条形图:
# barplot
palette = c("Grays","Light Grays","Blues2","Blues3","Purples2","Purples3","Reds2","Reds3","Greens2")
# loop
lapply(seq_along(unique(enrich$group)), function(x){
tmp <- enrich |> dplyr::filter(group == unique(enrich$group)[x]) |>
dplyr::arrange(desc(pvalue))
tmp$Description <- factor(tmp$Description,levels = tmp$Description)
# plot
p <-
ggplot(tmp) +
geom_col(aes(x = -log10(pvalue),y = Description,fill = -log10(pvalue)),
width = 0.75) +
geom_line(aes(x = log10(ratio),y = as.numeric(Description)),color = "grey50") +
geom_point(aes(x = log10(ratio),y = Description),size = 3,color = "orange") +
theme_bw() +
scale_y_discrete(position = "right",
labels = function(x) stringr::str_wrap(x, width = 40)) +
scale_x_continuous(sec.axis = sec_axis(~.,name = "log10(ratio)")) +
colorspace::scale_fill_binned_sequential(palette = palette[x]) +
ylab("")
return(p)
}) -> gglist
# assign names
names(gglist) <- paste("C",1:9,sep = "")插入到主图:
# insert bar plot
pdf('sc_ggplot_go.pdf',height = 20,width = 16,onefile = F)
visCluster(object = st.data,
plot.type = "both",
line.side = "left",
column_names_rot = 45,
markGenes = pbmc.markers$gene,
cluster.order = c(1:9),
ggplot.panel.arg = c(5,0.5,16,"grey90",NA),
gglist = gglist)
dev.off()
也许你可以插入其它相关的图形,让你的图更加丰富一些。比如拟时序分析的单个基因的趋势图等。大家可以自己试试。
4结尾
路漫漫其修远兮,吾将上下而求索。
欢迎加入生信交流群。加我微信我也拉你进 微信群聊 老俊俊生信交流群 (微信交流群需收取 20 元入群费用,一旦交费,拒不退还!(防止骗子和便于管理)) 。QQ 群可免费加入, 记得进群按格式修改备注哦。
声明:文中观点不代表本站立场。本文传送门:https://eyangzhen.com/239122.html


