
论文
Haplotype-resolved diverse human genomes and integrated analysis of structural variation
science.abf7117.pdf
论文里32个人的基因组,组装得到64套单倍型,论文里基于基因组比对的结构变异检测自己开发的流程pav
链接 https://github.com/EichlerLab/pav/tree/master
最近发表的Nature中国人的泛基因组论文里也提到
A pangenome reference of 36 Chinese populations
36人Pan_Nature.pdf
方法部分写到
We then used Phased Assembly Variant (PAV) caller v1.2 (ref. 17), which can prune duplicated alignments caused by minimap2, to detect small variants and SVs in each haplotype on the basis of the output CIGAR sequence from minimap2
这套流程具体的原理还是看不太明白,先把流程安装好,跑一下示例数据再说
安装
https://github.com/EichlerLab/pav/tree/master 先把这个下载下来,解压
然后把这个 https://github.com/EichlerLab/svpop/tree/master 下载下来
把svpoplib这个文件夹放到 pav-master文件下下
把 https://kgithub.com/paudano/kanapy/tree/master
这个也下载下来重命名为kanapy也放到 pav-master文件夹下
pip install matplotlib-venn 安装一下这个模块
其他的一些依赖我应该是都安装过了,比如minimap2 samtools这些
我的pav-master文件夹下有如下内容


然后参考 https://github.com/EichlerLab/pav/blob/master/EXAMPLE.md 这个链接运行一下示例数据,新建一个practice文件夹
文件夹下放两个文件
assemblies.tsv config.json

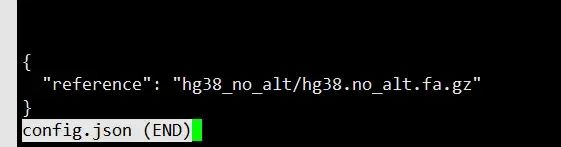
数据放到对应的文件夹下,运行命令
snakemake -s ../Snakefile --configfiles config.json --cores 32 -pn
snakemake -s ../Snakefile --configfiles config.json --cores 32 -p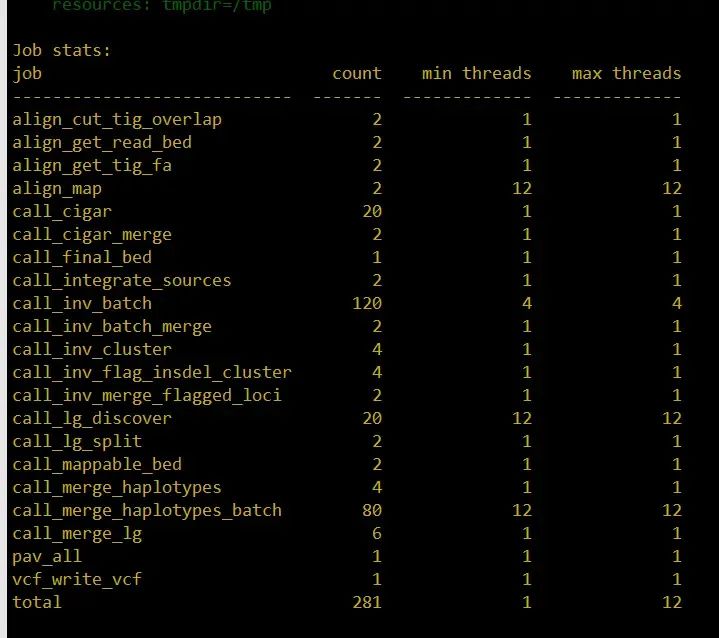
总共有这么多步骤
最终生成的结果怎么看还不知道
推文记录的是自己的学习笔记,大概率存在错误!
声明:文中观点不代表本站立场。本文传送门:https://eyangzhen.com/246807.html


