
整个流程是minimap2比对,然后是syri做变异检测,最后使用plotsr这个命令可视化展示结果,用到的命令是
plotsr --sr syri.out --genomes genomes.txt -W 4 -H 4 -o 3.pdf这个默认的图例是如下
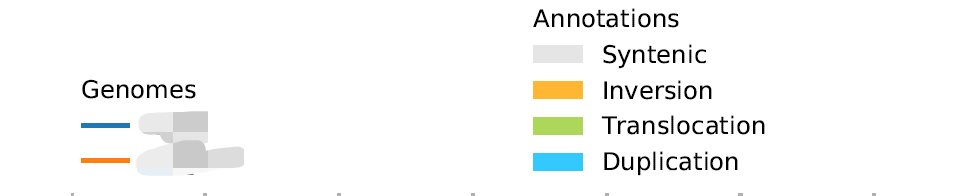

都是单列排布,注释那个图例四周空间还挺多的,我想把它改成两行两列的形式排布,查了一下plotsr的github主页
这里提到还有一个 –cfg参数可以提供一个配置文件修改一些细节,github主页提供了一个示例文件,这个文件的内容有
## COLOURS and transparency for alignments (syntenic, inverted, translocated, and duplicated)
syncol:#CCCCCC
invcol:#FFA500
tracol:#9ACD32
dupcol:#00BBFF
alpha:0.8
## Margins and dimensions:
chrmar:0.1 ## Adjusts the gap between chromosomes and tracks. Higher values leads to more gap
exmar:0.1 ## Extra margin at the top and bottom of plot area
## LEGEND
legend:T ## To plot legend use T, use F to not plot legend
genlegcol:-1 ## Number of columns for genome legend, set -1 for automatic setup
bbox:0,1.01,0.5,0.3 ## [Left edge, bottom edge, width, height]
bbox_v:0,1.1,0.5,0.3 ## For vertical chromosomes (using -v option)
bboxmar:0.5 ## Margin between genome and annotation legendslegend 设置为T或者F可以调节图例的有无
genlegcol 后面的数字设置的是Genomes那个图例的排布,单位是列,比如改成2,图例就变成了如下的样子
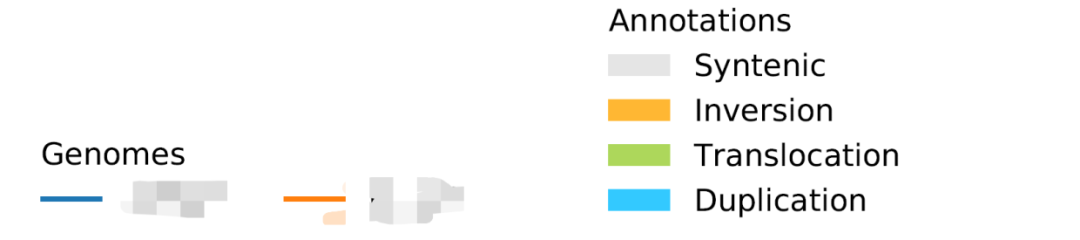
使用方式
plotsr --sr syri.out --genomes genomes.txt -W 4 -H 4 --cfg base.cfg -o 3.pdf但是没有找到调节Annotations这个图例的参数
看了一下plotsr的代码,python写的工具,大体上能够看懂哪是哪
plotsr.py 这个脚本里268行ncol参数是控制Annotations图例的列数的,我把这个1改成2就变成了2列显示

我是直接使用conda安装的这个工具,plotsr.py这个脚本存放的路径是
/home/myan/anaconda3/envs/syri/lib/python3.9/site-packages/plotsr/plotsr.py
拟南芥的数据试试
minimap2 -ax asm5 --eqx eri.fa c24.fa -o abc.sam -t 8
syri -c abc.sam -r eri.fa -q c24.fa -k -F S --nc 8
plotsr --sr syri.out --genomes genomes.txt -W 10 -H 4 -o abc.pdf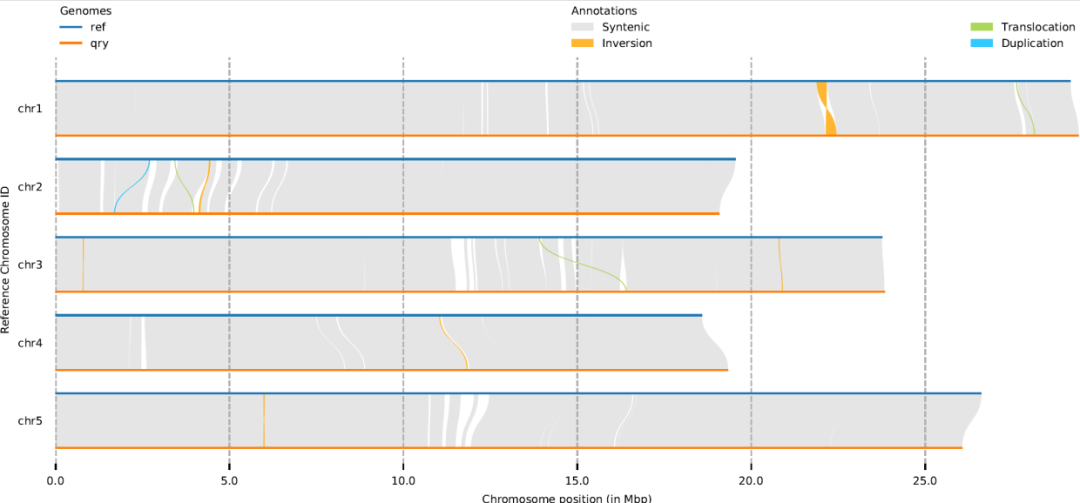
最后提供一个 minimap2+syri+plotsr分析的snakemake流程
SAMPLES, = glob_wildcards("../04.final.data/{sample}.fa")
print("Total sample size: ",len(SAMPLES))
rule all:
input:
expand("01.minimap2.syri/02.syri/{sample}/{sample}_1.pdf",sample=SAMPLES)
rule minimap2_sam:
input:
ref = "00.ref/T2T.fa",
qry = "00.qrys/{sample}.fa"
output:
"01.minimap2.syri/01.sam/{sample}.sam"
threads:
24
resources:
mem_mb = 100000
shell:
"""
minimap2 -ax asm5 --eqx {input.ref} {input.qry} -o {output} -t 24
"""
rule syri_call:
input:
ref = "00.ref/T2T.fa",
qry = "00.qrys/{sample}.fa",
sam = rules.minimap2_sam.output
output:
vcf = "01.minimap2.syri/02.syri/{sample}/{sample}_syri.vcf",
txt = "01.minimap2.syri/02.syri/{sample}/{sample}_syri.out"
threads:
24
resources:
mem_mb = 96000
params:
output_folder = "01.minimap2.syri/02.syri/{sample}",
prefix = "{sample}_"
shell:
"""
syri -c {input.sam} -r {input.ref} -q {input.qry} -k -F S --nc {threads} --dir {params.output_folder} --prefix {params.prefix}
"""
rule vim_genomes:
input:
ref = "00.ref/T2T.fa",
qry = "00.qrys/{sample}.fa",
vcf = rules.syri_call.output
output:
"01.minimap2.syri/02.syri/{sample}/genomes.txt"
threads:
2
resources:
mem_mb = 8000
params:
ref = "T2T",
qry = "{sample}"
run:
fw = open(output[0],'w')
fw.write("%st%sn%st%sn"%(input[0],params[0],input[1],params[1]))
fw.close()
rule plotsr:
input:
syri_out = rules.syri_call.output.txt,
genomes_txt = rules.vim_genomes.output
output:
"01.minimap2.syri/02.syri/{sample}/{sample}_1.pdf"
threads:
2
resources:
mem_mb = 8000
shell:
"""
plotsr --sr {input.syri_out} --genomes {input.genomes_txt} -W 4 -H 4 -o {output}
"""推文记录的是自己的学习笔记,内容可能会存在错误,请大家批判着看,欢迎大家指出其中的错误
示例数据和代码可以给推文点赞,然后点击在看,最后留言获取
声明:文中观点不代表本站立场。本文传送门:https://eyangzhen.com/28860.html


