
1引言
前面 ClusterGVis 可以支持对 monocle2 的时序热图: ClusterGVis 对接 monocle2 拟时序热图。但是官方对 monocle2 已经不再维护了,转而开发了 monocle3 来进行时序分析,但是之前的热图可视化的功能已经被弃用。如果你想用 3 的版本,又想画 2 版本的热图,貌似自己得费一点功夫才行,网上也有一些教程。
画热图的本质无非是根据你自己筛选出来的时序差异基因,然后提取对应的表达矩阵进行绘图即可,这里参考了网上的代码,封装一个 pre_pseudotime_matrix 函数来做这样的事情,返回一个目标基因的表达矩阵,然后你就可以给 ClusterGVis 来做一些可视化的事情了。
参考链接:
2安装
重新安装获取新功能:
# Note: please update your ComplexHeatmap to the latest version!
# install.packages("devtools")
devtools::install_github("junjunlab/ClusterGVis")3使用介绍
首先用 monocle3 进行拟时序分析,然后找到差异基因,这里就是包的示例代码了:
# devtools::install_github('cole-trapnell-lab/monocle3')
library(monocle3)
library(dplyr)
cell_metadata <- readRDS(system.file('extdata',
'worm_embryo/worm_embryo_coldata.rds',
package='monocle3'))
gene_metadata <- readRDS(system.file('extdata',
'worm_embryo/worm_embryo_rowdata.rds',
package='monocle3'))
expression_matrix <- readRDS(system.file('extdata',
'worm_embryo/worm_embryo_expression_matrix.rds',
package='monocle3'))
cds <- new_cell_data_set(expression_data=expression_matrix,
cell_metadata=cell_metadata,
gene_metadata=gene_metadata)
cds <- preprocess_cds(cds)
cds <- align_cds(cds, alignment_group =
"batch", residual_model_formula_str = "~ bg.300.loading +
bg.400.loading + bg.500.1.loading + bg.500.2.loading +
bg.r17.loading + bg.b01.loading + bg.b02.loading")
cds <- reduce_dimension(cds)
cds <- cluster_cells(cds)
cds <- learn_graph(cds)
cds <- order_cells(cds, root_pr_nodes='Y_21')
modulated_genes <- graph_test(cds, neighbor_graph = "principal_graph", cores = 4)
genes <- row.names(subset(modulated_genes, q_value == 0 & morans_I > 0.25))然后就是提取差异基因的表达矩阵:
mat <- pre_pseudotime_matrix(cds_obj = cds,
gene_list = genes)
# check
head(mat[1:5,1:5])
# 1 2 3 4 5
# WBGene00013882 -0.7265393 -0.7263892 -0.7262392 -0.7260891 -0.7259390
# WBGene00002085 0.8059370 0.8076543 0.8093716 0.8110889 0.8128062
# WBGene00016114 2.8256183 2.8198074 2.8139964 2.8081855 2.8023746
# WBGene00012753 2.7917688 2.7861478 2.7805268 2.7749058 2.7692848
# WBGene00015354 -1.1233255 -1.1229564 -1.1225872 -1.1222180 -1.1218489聚类,绘图:
library(ClusterGVis)
# kmeans
ck <- clusterData(exp = mat,
cluster.method = "kmeans",
cluster.num = 5)
# add line annotation
pdf('monocle3.pdf',height = 10,width = 8,onefile = F)
visCluster(object = ck,
plot.type = "both",
add.sampleanno = F,
markGenes = sample(rownames(mat),30,replace = F))
dev.off()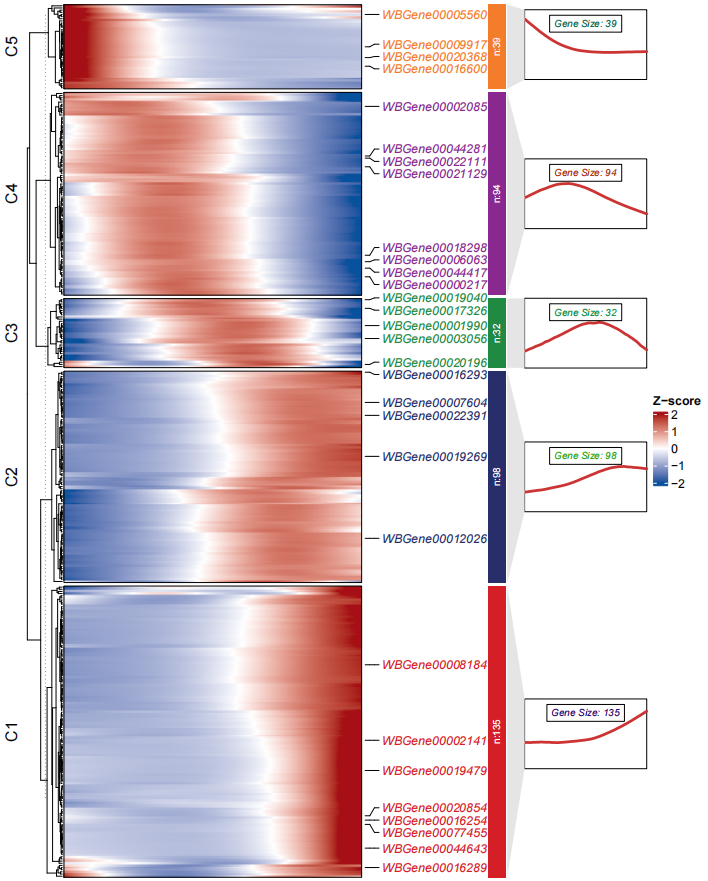

富集分析,添加注释通路什么的参考使用见 **https://github.com/junjunlab/ClusterGVis**。
4结尾
路漫漫其修远兮,吾将上下而求索。
欢迎加入生信交流群。加我微信我也拉你进 微信群聊 老俊俊生信交流群 (微信交流群需收取 20 元入群费用,一旦交费,拒不退还!(防止骗子和便于管理)) 。QQ 群可免费加入, 记得进群按格式修改备注哦。
声明:文中观点不代表本站立场。本文传送门:https://eyangzhen.com/29429.html
