
我做的是植物,首先是使用RepeatModeler构建自己物种的重复序列数据库
BuildDatabase -name ABC ABC.genome.fasta
RepeatModeler -database ABC -pa 24 -LTRStruct 1>repeatmodeler.log 2>&1这一步生成的AAA-families.fa 文件里有很多Unknown


然后是用RepeatMasker做重复序列的注释
RepeatMasker -e rmblast -pa 24 -qq -xsmall -lib AAA-families.fa AAA.genome.fasta 1>repeatmasker.log 2>&1这一步生成的.tbl文件里未分类的达到30%多

我用到的RepeatModeler和RepeatMasker都是用conda安装的,没有进行额外的配置
我去翻了翻第一步RepeatModeler的运行日志文件,发现里面是有一个报错的
Running Ltr_retriever.../home/myan/anaconda3/envs/repeat/bin/faToTwoBit: error while loading shared libraries: libssl.so.1.0.0: cannot open shared object file: No such file or directory应该是Ltr_retriever没有运行成功,但是整个程序是运行成功了
我试了一下我EDTA环境下的Ltr_retrirver是可以运行成功的,所以手动配置了RepeatModeler和RepeatMasker,依赖软件用到的是EDTA环境下的。这里RepeatMasker是4.1.5,Dfam库的序列条数多了很多
这次再运行完两个流程未分类的占到15%左右,上面提到的未分类过多的应该就是Ltr_retriever没有运行成功导致的
这次生成的AAA-families.fa 文件里还有一些是未分类的Unknown
这个主要是两类,一类是LTR/Unknown

另外一类是彻底的Unknown
首先对LTR/Unknown去做分类,最开始用到的是TEsorter
我用到的是conda安装,运行的命令是
TEsorter LTRandUnknown.fa基本没作用,可能是我运行的有问题吧
然后又尝试了DeepTE
但是有一个问题是LTR会被分类成其他类型的转座子,这里我只保留被分类成LTR的
import sys
input_file = sys.argv[1]
output_file = sys.argv[2]
fr = open(input_file,'r')
fw = open(output_file,'w')
for line in fr:
if len(line.strip().split("t")[1].split("_")) == 3 and line.strip().split("t")[1].split("_")[1] == "LTR":
prefix = line.strip().split("t")[0].split("#")[0]
suffix = "/".join(line.strip().split("t")[1].split("_")[1:3])
fw.write("%st%sn"%(line.strip().split("t")[0],prefix+"#"+suffix))
#print(i)
fw.close()
fr.close()用这个脚本去处理opt_DeepTE.txt文件得到
python 001.py opt_DeepTE.txt unknown_id_dict.txtid对照表
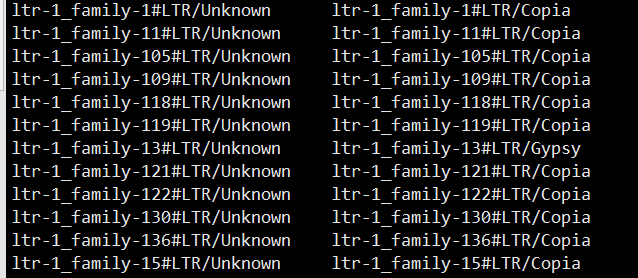
第二类是彻底的unknown的也用DeepTE去分类,然后只保留LTR和DNA的类型,因为被分为其他类型的我暂时不知道怎么在AAA-families.fa里更改名字
同样的用上面的脚本处理opt_DeepTE.txt文件得到LTR的id对照表
然后再用下面的脚本处理opt_DeepTE.txt文件得到DNA的id对照表
import sys
input_file = sys.argv[1]
output_file = sys.argv[2]
fr = open(input_file,'r')
fw = open(output_file,'w')
for line in fr:
if len(line.strip().split("t")[1].split("_")) >= 2 and line.strip().split("t")[1].split("_")[1] == "DNA":
prefix = line.strip().split("t")[0].split("#")[0]
suffix = "DNA/"+"-".join(line.strip().split("t")[1].split("_")[2:])
fw.write("%st%sn"%(line.strip().split("t")[0],prefix+"#"+suffix))
#print(i)
fw.close()
fr.close()python 002.py ABC/opt_DeepTE.txt DNA_unknown_id_dict.txt最后将三个id对照表合并,用如下脚本去替换AAA-families.fa中的id
import sys
input_fa = sys.argv[1]
input_dict = sys.argv[2]
output_fa = sys.argv[3]
fr01 = open(input_fa,'r')
fr02 = open(input_dict,'r')
id_dict = {}
for line in fr02:
id_dict[line.strip().split("t")[0]] = line.strip().split("t")[1]
fw = open(output_fa,'w')
for line in fr01:
if line.startswith(">") and line.strip().split(" ")[0].replace(">","") in id_dict.keys():
first_part = id_dict[line.strip().split(" ")[0].replace(">","")]
second_part = ' '.join(line.strip().split(" ")[1:])
fw.write(">"+first_part+" "+second_part+"n")
else:
fw.write(line)
fr01.close()
fr02.close()
fw.close()python 003.py AAA-families.fa unknown_id_dict.txt AAA-families01.fa然后用AAA-families01.fa去做repeatMasker,最终未分类的比例降到了5%左右
推文记录的是自己的学习笔记,内容可能会存在错误,请大家批判着看,欢迎大家指出其中的错误
声明:文中观点不代表本站立场。本文传送门:https://eyangzhen.com/36307.html


