
论文
De novo assembly, annotation, and comparative analysis of 26 diverse maize genomes
玉米science.pdf
玉米science补充材料.pdf
数据代码链接
github
因为玉米的数据太大了,这里我用拟南芥的数据,拟南芥的数据来源于论文
PRJEB31147
根据Project ID获取所有的SRA number
esearch -db sra -query PRJEB31147 | efetch -format runinfo | grep 'PACBIO' | awk -F "," '{print $1","$12}'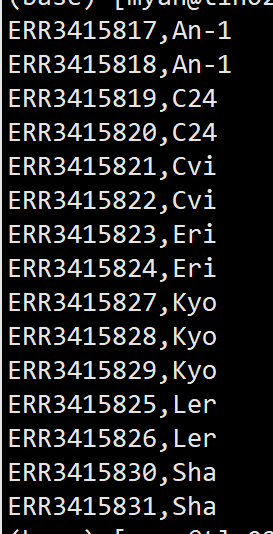

每个样本都有两个数据,还有一个样本是3个数据,每个样本只下载一个数据
用ffq这个工具根据SRA id获取ftp下载链接
ffq ERR3415817ftp://ftp.sra.ebi.ac.uk/vol1/fastq/ERR341/007/ERR3415817/ERR3415817_subreads.fastq.gz
ftp://ftp.sra.ebi.ac.uk/vol1/fastq/ERR341/009/ERR3415819/ERR3415819_subreads.fastq.gz
ftp://ftp.sra.ebi.ac.uk/vol1/fastq/ERR341/001/ERR3415821/ERR3415821_subreads.fastq.gz
ftp://ftp.sra.ebi.ac.uk/vol1/fastq/ERR341/003/ERR3415823/ERR3415823_subreads.fastq.gz
ftp://ftp.sra.ebi.ac.uk/vol1/fastq/ERR341/007/ERR3415827/ERR3415827_subreads.fastq.gz
ftp://ftp.sra.ebi.ac.uk/vol1/fastq/ERR341/005/ERR3415825/ERR3415825_subreads.fastq.gz
ftp://ftp.sra.ebi.ac.uk/vol1/fastq/ERR341/000/ERR3415830/ERR3415830_subreads.fastq.gz获取到链接地址以后在本地开着科学上网工具下载,暂时想不到其他比较好的下载方法了
science论文里会把每个fq/fa进行拆分,分别比对,然后再合并bam文件,这个拟南芥的数据量也不大,直接比对吧,不拆分了,science论文里用到的拆分脚本是 fasta-splitter.pl
下载有点慢 就下了三个数据
比对用到到的是ngmlr,安装直接用conda
ngmlr --reference TAIR10_chr_all.fas --query ../pacbio.fq/ERR3415817_subreads.fastq.gz --presets pacbio --output An_1.sam --bam-fix --no-progress --threads 8
ngmlr --reference TAIR10_chr_all.fas --query ../pacbio.fq/ERR3415821_subreads.fastq.gz --presets pacbio --output Cvi.sam --bam-fix --no-progress --threads 8
ngmlr --reference TAIR10_chr_all.fas --query ../pacbio.fq/ERR3415823_subreads.fastq.gz --presets pacbio --output Eri.sam --bam-fix --no-progress --threads 8这个速度很慢 一个样本需要运行2个小时左右
这个输出的sam文件用samtools操作的的时候还报错了,暂时不知道什么原因,把数据减小,重新比对下试试
zcat ERR3415817_subreads.fastq.gz | seqkit sample -p 0.2 -o An_1.fq
ngmlr --reference TAIR10_chr_all.fas --query ../pacbio.fq/An_1.fq --output An_1p2.sam --threads 8
samtools sort -@ 8 -o An_1p2.sorted.bam An_1p2.sam报错
samtools sort: truncated file. Aborting暂时没有查到是什么原因,换minimap2软件比对试试
minimap2 -ax map-pb TAIR10_chr_all.fas ../pacbio.fq/An_1.fq > An_1.samsamtools sort -@ 4 -o An_1.sorted.bam An_1.sam这个就没有遇到报错
samtools view -@ 4 -b -o An_1.bam An_1.samsam转bam也没问题
以上流程写了一个snakemake
SAMPLES, = glob_wildcards("{sample}_subreads.fastq.gz")
print(SAMPLES)
rule all:
input:
#expand("{sample}.sam",sample=SAMPLES)
expand("{sample}.sorted.bam.bai",sample=SAMPLES)
rule b_minimap2:
input:
fq = "{sample}_subreads.fastq.gz",
fa = "TAIR10_chr_all.fas"
output:
"{sample}.sam"
threads:
4
shell:
"""
minimap2 -ax map-pb {input.fa} {input.fq} > {output}
"""
#https://github.com/HuffordLab/NAM-genomes/blob/master/structural-variation/sv-calling/runSAMcleaner.sh
rule d_samtools_view:
input:
rules.b_minimap2.output
output:
"{sample}.bam"
threads:
4
shell:
"""
samtools view -@ {threads} -b -o {output} {input}
"""
rule e_samtools_sort:
input:
rules.d_samtools_view.output
output:
"{sample}.sorted.bam"
threads:
4
shell:
"""
samtools sort -@ {threads} -o {output} {input}
"""
rule f_samtools_index:
input:
rules.e_samtools_sort.output
output:
"{sample}.sorted.bam.bai"
threads:
4
shell:
"""
samtools index {input}
"""比对好以后用sniffles
使用conda安装
conda install sniffles这个最新的已经是2点几的版本了,science论文里用到的是1点几的版本,很多参数已经不一样了
2.几版本用到的命令是
sniffles --input ERR3415823.sorted.bam --snf ERR3415823.snf --threads 12
sniffles --input ERR3415821.sorted.bam --snf ERR3415821.snf --threads 12
sniffles --input ERR3415817.sorted.bam --snf ERR3415817.snf --threads 12
sniffles --input ERR3415817.snf ERR3415821.snf ERR3415823.snf --vcf multisample.vcf
sniffles --input ERR3415817.sorted.bam --genotype-vcf multisample.vcf --vcf ERR3415817_genotypes.vcf这个运行速度很快
还有一个问题是 这个多样本的vcf文件里每个sample里是有GT 信息的,那么还需要单独再运行最后一个命令吗?
看下sniffles的论文本帮助文档
单独安装一个和science论文里一样版本的sniffles 1.0.11 现在安装的是2.0.7
conda install sniffles=1.0.11 -y这个安装在了sv这个环境里
sniffles --threads 12 --mapped_reads ERR3415817.sorted.bam --max_distance 1000 --min_support 10 --min_length 100 --min_seq_size 5000 --vcf ERR3415817_r1.vcf报错
No MD string detected! Check bam file! Otherwise generate using e.g. samtools.在使用minimap2比对的时候需要加参数
用samtools
samtools calmd -b ERR3415817.sorted.bam TAIR10_chr_all.fas > abc.sorted.ba
sniffles --threads 12 --mapped_reads abc.sorted.bam --max_distance 1000 --min_support 10 --min_length 100 --min_seq_size 5000 --vcf ERR3415817_r1.vcf这样是可以得到结果的
查samtools的帮助文档的时候
http://www.htslib.org/doc/samtools-calmd.html
运行这个命令
samtools calmd -bAr aln.bam > aln.baq.bam是有报错的 samtools calmd: BAQ alignment failed: Numerical result out of range 暂时不理解这些参数的意思
可以参考这个链接 https://www.jianshu.com/p/68f6e35fa4a2 有时间仔细看看
samtools calmd -b ERR3415821.sorted.bam TAIR10_chr_all.fas > sample21.sorted.bam
samtools calmd -b ERR3415823.sorted.bam TAIR10_chr_all.fas > sample23.sorted.bam
sniffles --threads 12 --mapped_reads sample21.sorted.bam --max_distance 1000 --min_support 10 --min_length 100 --min_seq_size 5000 --vcf ERR3415821_r1.vcf
sniffles --threads 12 --mapped_reads sample23.sorted.bam --max_distance 1000 --min_support 10 --min_length 100 --min_seq_size 5000 --vcf ERR3415823_r1.vcf合并第一轮的vcf
ls *_r1.vcf > vcf.fofn
SURVIVOR merge vcf.fofn 1000 1 1 -1 -1 -1 survivor_merged_1kbdist_r1.vcf运行第二轮的sniffles
sniffles --threads 12 --mapped_reads abc.sorted.bam --Ivcf survivor_merged_1kbdist_r1.vcf --vcf sample17_r2.vcf
sniffles --threads 12 --mapped_reads sample21.sorted.bam --Ivcf survivor_merged_1kbdist_r1.vcf --vcf sample21_r2.vcf
sniffles --threads 12 --mapped_reads sample23.sorted.bam --Ivcf survivor_merged_1kbdist_r1.vcf --vcf sample23_r2.vcf合并
ls sample*_r2.vcf > vcf_r2.fofn
SURVIVOR merge vcf_r2.fofn 1000 -1 1 -1 -1 -1 survivor_merged_1kbdist_r2.vcf第二轮合并后的vcf和第一轮合并的vcf有啥区别暂时看不出来
欢迎大家关注我的公众号
小明的数据分析笔记本
声明:文中观点不代表本站立场。本文传送门:https://eyangzhen.com/156011.html
