
1引言
Deeptools 是目前比较流行的可视化软件,除此之外还有一款 R 包 EnrichedHeatmap, 不过感觉用的人似乎并不多,之前我也介绍过,这次我们拿数据实战一下。
安装:
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("EnrichedHeatmap")数据:
在 GSE205706 上随便下载了一些 bigwig 文件:

2示例
首先准备一下基因的 TSS 数据 (你可以从 GTF 或者 UCSC 获取基因的位置信息的 bed 文件),使用 promoters函数提取 TSS 位点信息:
library(EnrichedHeatmap)
library(tidyverse)
library(GenomicRanges)
library(rtracklayer)
library(circlize)
hg38.bed <- read.delim("hg38.bed",header = F) |>
mutate(seqnames = V1,start = V2,end = V3,strand = V6) |>
select(seqnames,start,end,strand) |> head(1000) |> GRanges()
tss <- promoters(hg38.bed,upstream = 0, downstream = 1)
tss
# GRanges object with 1000 ranges and 0 metadata columns:
# seqnames ranges strand
# <Rle> <IRanges> <Rle>
# [1] chr1 11868 +
# [2] chr1 12009 +bigwig 文件准备:
bw.files <- c("GSM6219545_K562-b1-Control-Input-r1_ChIP.bigWig",
"GSM6219547_K562-b1-RBFOX2-KD-Input-r1_ChIP.bigWig",
"GSM6219541_K562-b1-Control-H3K4me3-r1_ChIP.bigWig",
"GSM6219543_K562-b1-RBFOX2-KD-H3K4me3-r1_ChIP.bigWig",
"GSM6219549_K562-b2-Control-SUZ12-r1_ChIP.bigWig",
"GSM6219557_K562-b3-Control-H3K27me3-r1_ChIP.bigWig",
"GSM6219565_K562-b4-Control-YTHDC1-r1_ChIP.bigWig")
sample <- c("Control","RBFOX2-KD","Control-H3K4me3","RBFOX2-KD-H3K4me3",
"Control-SUZ12","Control-H3K27me3","Control-YTHDC1")
bg_col <- RColorBrewer::brewer.pal(7, "Set2")批量计算在 TSS 位置处的信号强度矩阵:
ht_list <- lapply(seq_along(bw.files), function(x){
bw <- import.bw(bw.files[x])
mat_trim = normalizeToMatrix(bw, genes, value_column = "score",
extend = 5000, mean_mode = "w0", w = 50,
# keep = c(0, 0.99),
background = 0, smooth = TRUE)
col_fun = colorRamp2(quantile(mat_trim, c(0, 0.99)), c("grey95", bg_col[x]))
# col_fun = colorRamp2(c(0,30), c("grey95", bg_col[x]))
# plot
ht <-
EnrichedHeatmap(mat_trim, col = col_fun, name = sample[x],
column_title = sample[x],
column_title_gp = gpar(fontsize = 10,
fill = bg_col[x]),
# top_annotation = HeatmapAnnotation(
# enriched = anno_enriched(
# ylim = c(0, 30))),
use_raster = F,
# show_heatmap_legend = lgd[x],
show_heatmap_legend = T
)
return(ht)
})最后绘图保存:
# merge plot list
pdf("ht.pdf",width = 15,height = 8,onefile = F)
draw(Reduce("+",ht_list), ht_gap = unit(0.8, "cm"),
heatmap_legend_side = "bottom")
dev.off()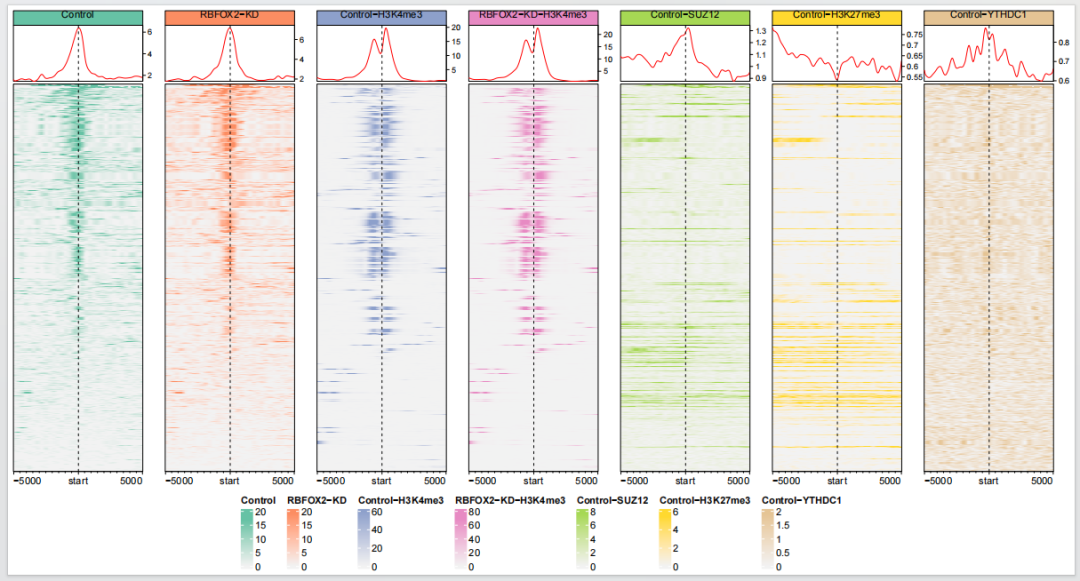
你可以看到这里的每个图的纵坐标都不一样,你也可以设置为一样的,这样可以展示不同条件下信号强度的差异:
lgd <- rep(c("T","F"),c(1,6))
# loop plot
ht_list2 <- lapply(seq_along(bw.files), function(x){
bw <- import.bw(bw.files[x])
mat_trim = normalizeToMatrix(bw, tss, value_column = "score",
extend = 5000, mean_mode = "w0", w = 50,
# keep = c(0, 0.99),
background = 0, smooth = TRUE)
# col_fun = colorRamp2(quantile(mat_trim, c(0, 0.99)), c("grey95", bg_col[x]))
col_fun = colorRamp2(c(0,30), c("grey95", bg_col[x]))
# plot
ht <-
EnrichedHeatmap(mat_trim, col = col_fun, name = sample[x],
column_title = sample[x],
column_title_gp = gpar(fontsize = 10,
fill = bg_col[x]),
top_annotation = HeatmapAnnotation(
enriched = anno_enriched(
ylim = c(0, 30))),
use_raster = F,
show_heatmap_legend = lgd[x],
# show_heatmap_legend = T
)
return(ht)
})
# merge plot list
pdf("ht_scale_y.pdf",width = 15,height = 6,onefile = F)
draw(Reduce("+",ht_list2), ht_gap = unit(0.8, "cm"),
heatmap_legend_side = "right")
dev.off()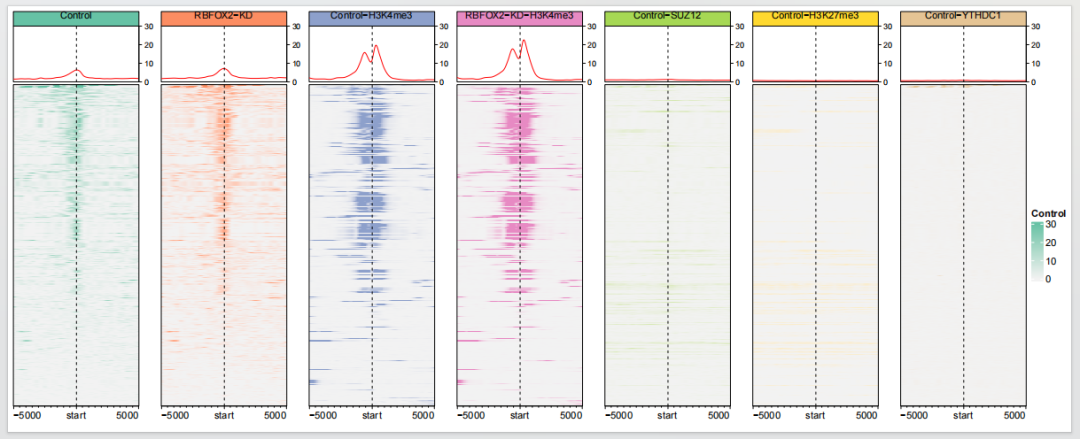
3生物学重复样本处理
有时候你可能有多个生物学重复样本,也许你想把它们合并,我们先看看各自的图形:
rep_file <- c("GSM6219545_K562-b1-Control-Input-r1_ChIP.bigWig",
"GSM6219546_K562-b1-Control-Input-r2_ChIP.bigWig",
"GSM6219541_K562-b1-Control-H3K4me3-r1_ChIP.bigWig",
"GSM6219542_K562-b1-Control-H3K4me3-r2_ChIP.bigWig")
sample <- c("Input-r1","Input-r2","H3K4me3-r1","H3K4me3-r2")
lgd <- rep(c("T","F"),c(1,3))
# loop
# x = 1
lapply(1:4, function(x){
bw <- import.bw(rep_file[x])
mat <- normalizeToMatrix(bw, tss,
value_column = "score",
extend = 5000, mean_mode = "w0", w = 50,
background = 0, smooth = TRUE)
# col_fun = colorRamp2(quantile(mat, c(0, 0.99)), c("grey95", bg_col[x]))
col_fun = colorRamp2(c(0,20), c("grey95", bg_col[x]))
# plot
ht <-
EnrichedHeatmap(mat, col = col_fun, name = sample[x],
column_title = sample[x],
column_title_gp = gpar(fontsize = 10,
fill = bg_col[x]),
top_annotation = HeatmapAnnotation(
enriched = anno_enriched(
ylim = c(0, 20))),
use_raster = F,
show_heatmap_legend = lgd[x],
# show_heatmap_legend = T
)
return(ht)
}) -> ht_list4
# merge plot list
pdf("ht_rep_samples.pdf",width = 8,height = 6,onefile = F)
draw(Reduce("+",ht_list4), ht_gap = unit(0.8, "cm"),
heatmap_legend_side = "right")
dev.off()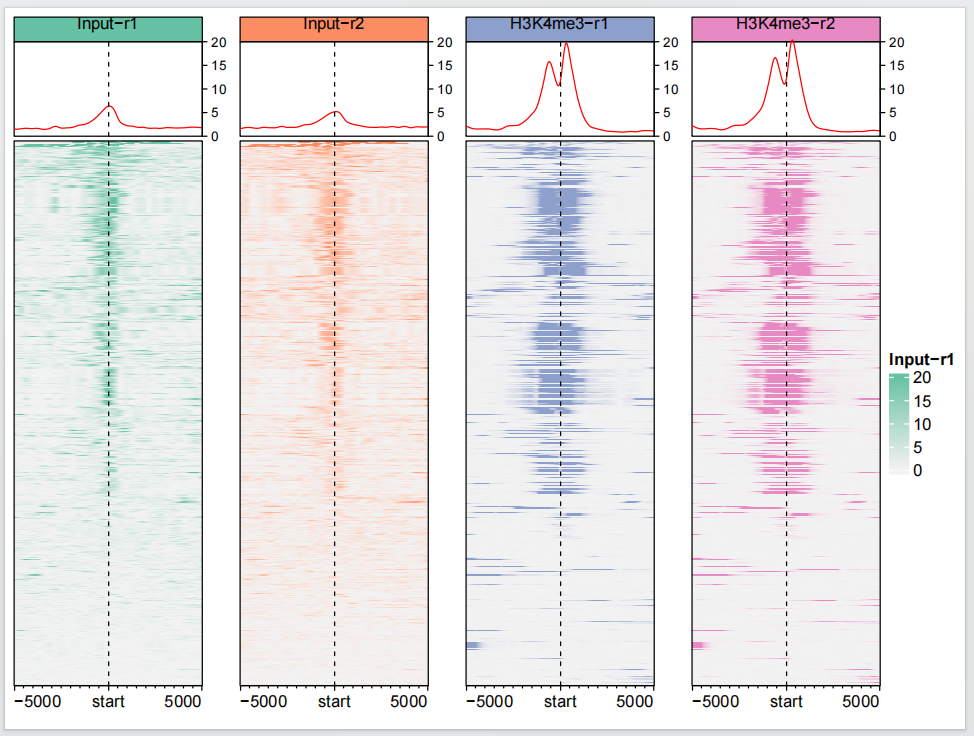
EnrichedHeatmap 提供了合并生物学重复的方法,取均值:
rep_file <- c("GSM6219545_K562-b1-Control-Input-r1_ChIP.bigWig",
"GSM6219546_K562-b1-Control-Input-r2_ChIP.bigWig",
"GSM6219541_K562-b1-Control-H3K4me3-r1_ChIP.bigWig",
"GSM6219542_K562-b1-Control-H3K4me3-r2_ChIP.bigWig")
sample <- c("Control-Input","Control-H3K4me3")
# loop
# x = 1
lapply(1:2, function(x){
rep_list <- lapply(c(x,x+1), function(x){
bw <- import.bw(rep_file[x])
})
mat_list = NULL
for(i in seq_along(rep_list)) {
mat_list[[i]] = normalizeToMatrix(rep_list[[i]], tss,
value_column = "score",
extend = 5000, mean_mode = "w0", w = 50,
background = 0, smooth = TRUE)
}
# get mean values
mat_mean = getSignalsFromList(mat_list)
# col_fun = colorRamp2(quantile(mat_mean, c(0, 0.99)), c("grey95", bg_col[x]))
col_fun = colorRamp2(c(0,30), c("grey95", bg_col[x]))
# plot
ht <-
EnrichedHeatmap(mat_mean, col = col_fun, name = sample[x],
column_title = sample[x],
column_title_gp = gpar(fontsize = 10,
fill = bg_col[x]),
top_annotation = HeatmapAnnotation(
enriched = anno_enriched(
ylim = c(0, 30))),
use_raster = F,
# show_heatmap_legend = lgd[x],
show_heatmap_legend = T
)
return(ht)
}) -> ht_list3
# merge plot list
pdf("ht_rep_mean.pdf",width = 6,height = 6,onefile = F)
draw(Reduce("+",ht_list3), ht_gap = unit(0.8, "cm"),
heatmap_legend_side = "right")
dev.off()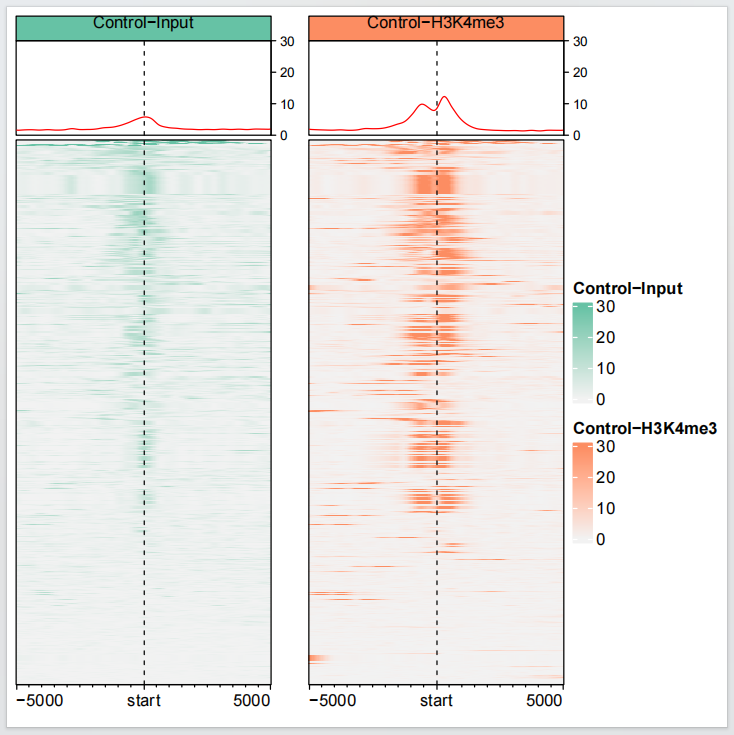
4分类
有时候我们希望给我们鉴定到的结合位点进行分类,然后进行可视化,下面展示一个例子:
rep_file <- c("GSM6219545_K562-b1-Control-Input-r1_ChIP.bigWig",
"GSM6219546_K562-b1-Control-Input-r2_ChIP.bigWig",
"GSM6219541_K562-b1-Control-H3K4me3-r1_ChIP.bigWig",
"GSM6219542_K562-b1-Control-H3K4me3-r2_ChIP.bigWig")
sample <- c("Input-r1","Input-r2","H3K4me3-r1","H3K4me3-r2")
lgd <- rep(c("T","F"),c(1,3))
row_split = sample(c("A", "B","C"), length(tss), replace = TRUE)
# loop
# x = 1
lapply(1:4, function(x){
bw <- import.bw(rep_file[x])
mat <- normalizeToMatrix(bw, tss,
value_column = "score",
extend = 5000, mean_mode = "w0", w = 50,
background = 0, smooth = TRUE)
# col_fun = colorRamp2(quantile(mat, c(0, 0.99)), c("grey95", bg_col[x]))
col_fun = colorRamp2(c(0,25), c("grey95", bg_col[x]))
# plot
ht <-
EnrichedHeatmap(mat, col = col_fun, name = sample[x],
column_title = sample[x],
column_title_gp = gpar(fontsize = 10,
fill = bg_col[x]),
top_annotation = HeatmapAnnotation(
lines = anno_enriched(gp = gpar(col = 2:4),ylim = c(0, 25))
),
use_raster = F,
# row_split = row_split,
# show_heatmap_legend = lgd[x],
show_heatmap_legend = T
)
return(ht)
}) -> ht_list5
# merge plot list
pdf("ht_rep_samples_clusters.pdf",width = 8,height = 6,onefile = F)
draw(Heatmap(row_split, col = structure(2:4, names = c("A","B","C")),
name = "cluster",
show_row_names = FALSE, width = unit(3, "mm")) +
Reduce("+",ht_list5),
ht_gap = unit(c(0.1,0.8,0.8,0.8), "cm"),
heatmap_legend_side = "bottom",
split = row_split)
dev.off()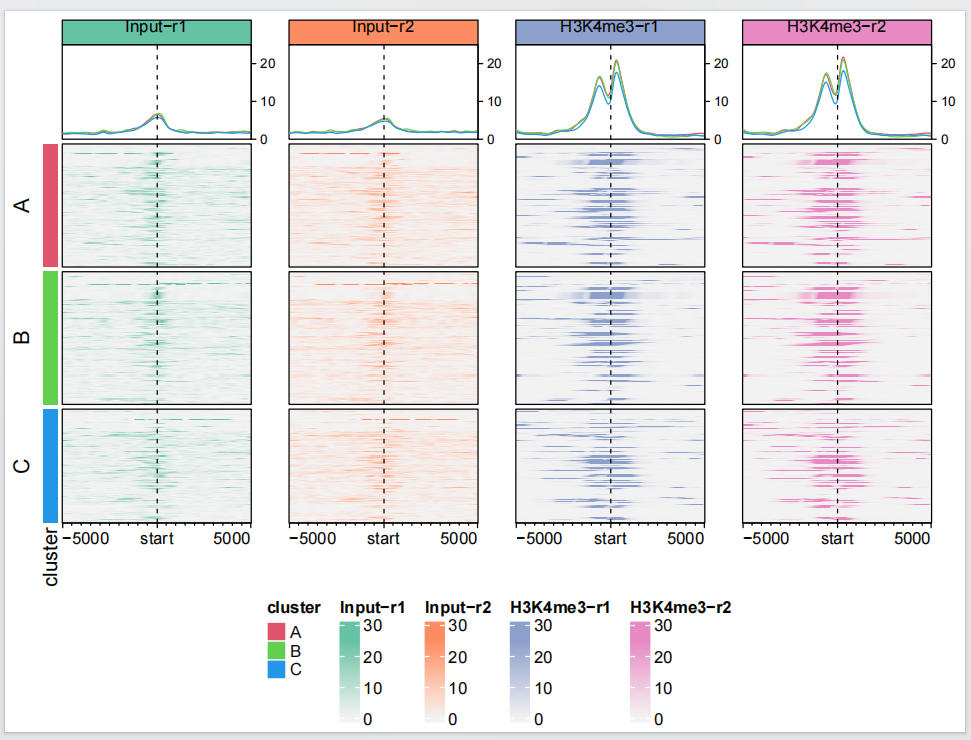
5提取 profile 图
有时候可能你只想展示 profile 图形,下面展示了怎么从已绘制的图形中提取出来:
# extract profile
htlist <- draw(Heatmap(row_split, col = structure(2:4, names = c("A","B","C")),
name = "cluster",
show_row_names = FALSE, width = unit(3, "mm")) +
Reduce("+",ht_list5),
ht_gap = unit(c(0.1,0.8,0.8,0.8), "cm"),
heatmap_legend_side = "bottom",
split = row_split)
# extract profile
add_anno_enriched = function(ht_list, name, ri, ci) {
pushViewport(viewport(layout.pos.row = ri, layout.pos.col = ci))
extract_anno_enriched(ht_list, name, newpage = FALSE)
upViewport()
}
# save
pdf("profile.pdf",width = 16,height = 4,onefile = F)
grid.newpage()
pushViewport(viewport(layout = grid.layout(nr = 1, nc = 4)))
add_anno_enriched(htlist,2 ,1, 1)
add_anno_enriched(htlist,3 ,1, 2)
add_anno_enriched(htlist,4 ,1, 3)
add_anno_enriched(htlist,5 ,1, 4)
upViewport()
dev.off()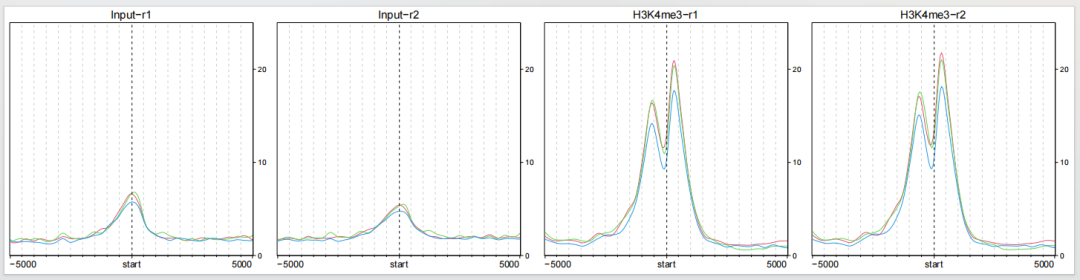
6提取数据 ggplot 绘图
如果你对上面的图形不太满意,甚至你可以自己提取数据进行绘图,这里自己写了一些简单的代码进行可视化:
profile:
library(ggplot2)
library(ggh4x)
library(colorspace)
# loop extract profile data
purrr::map_df(seq_along(ht_list5),function(x){
norm_mat <- ht_list5[[x]]@matrix
# =====================
# extract profile data
# group for rows
# x = "A"
purrr::map_df(sort(unique(row_split)),function(split){
idx <- base::grep(split,row_split)
mean_mat <- apply(norm_mat[idx,], 2, mean)
# profile to df
mat_df <- data.frame(density = mean_mat,
x = seq(-5000,5000,length.out = length(mean_mat)),
sample = ht_list5[[x]]@name,
split = split)
return(mat_df)
}) -> df.profile
return(df.profile)
}) -> df.profile
# plot
ggplot(df.profile) +
geom_line(aes(x = x,y = density,color = split)) +
theme_bw() +
theme(panel.grid = element_blank()) +
facet_wrap(~sample,nrow = 1) +
xlab("") +
scale_color_brewer(palette = "Set1")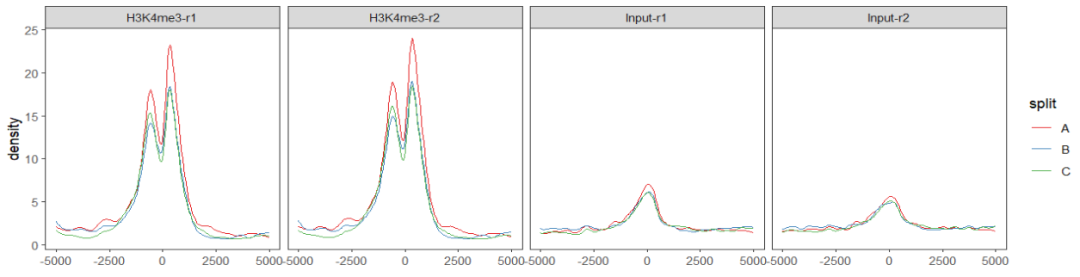
heatmap:
# loop extract heatmap data
# x = 1
purrr::map_df(seq_along(ht_list5),function(x){
norm_mat <- ht_list5[[x]]@matrix
# =====================
# extract heatmap data
purrr::map_df(sort(unique(row_split)),function(split){
idx <- base::grep(split,row_split)
tmp_mat <- data.frame(norm_mat[idx,])
colnames(tmp_mat) <- seq(-5000,5000,length.out = ncol(tmp_mat))
tmp_mat$y <- rownames(tmp_mat)
# tmp_mat$rowsum <- rowSums(tmp_mat)
df.long <- reshape2::melt(tmp_mat,id.vars = "y")
colnames(df.long)[2] <- c("x")
df.long$split <- split
df.long$sample <- ht_list5[[x]]@name
df.long$x <- as.numeric(unfactor(df.long$x))
df.long$y <- as.numeric(df.long$y)
return(df.long)
}) -> df.heatmap
# deal with extreme values
q99 <- quantile(df.heatmap$value,c(0,0.99))
df.heatmap.new <- df.heatmap |>
mutate(value = ifelse(value > q99[2],q99[2],value))
# orders
ht.sm <- df.heatmap |>
group_by(split,y) |>
summarise(rowsum = sum(value)) |>
arrange(split,rowsum) |> ungroup()
# # assign orders for each row split
# sp = "A"
map_df(unique(row_split),function(sp){
tmp.sm <- subset(ht.sm,split == sp)
tmp.heatmap <- subset(df.heatmap.new,split == sp)
tmp.heatmap$y <- factor(tmp.heatmap$y,levels = tmp.sm$y)
return(tmp.heatmap)
}) -> df.heatmap.final
return(df.heatmap.final)
}) -> df.heatmap
# plot
ggplot(df.heatmap) +
geom_raster(aes(x = x,y = y,fill = value)) +
theme_bw() +
theme(panel.grid = element_blank()) +
facet_grid2(split~sample,switch = "y") +
xlab("") + ylab("") +
scale_fill_continuous_sequential(palette = "Reds 3") +
coord_cartesian(expand = 0) +
theme(strip.placement = "outside",
axis.ticks.y = element_blank(),
axis.text.y = element_blank(),
panel.spacing.x = unit(0.6,"cm"),
axis.text.x = element_text(angle = 45,hjust = 1))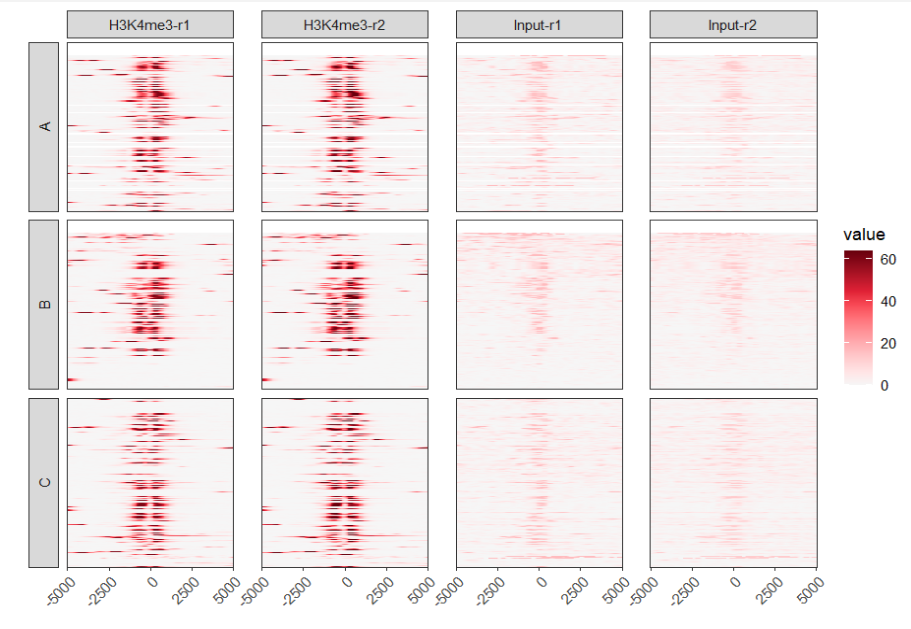
你也可以使用 aplot 把 profile 和 heatmap 拼起来,下面是个简单示例,具体细节你可以自己进一步调整:
# aplot plots
sp <- unique(df.profile$sample)
# loop plot
lapply(sp, function(s){
ptop <- ggplot(df.profile |> dplyr::filter(sample %in% s)) +
geom_line(aes(x = x,y = density,color = split)) +
theme_bw() +
theme(panel.grid = element_blank(),
axis.text.x = element_blank(),
plot.margin = margin(t = 0.2,r = .2,b = -0.2,l = .2,
unit = "cm"),
axis.title.x = element_blank(),
axis.ticks.x = element_blank()) +
facet_wrap(~sample,nrow = 1) +
xlab("") +
scale_color_brewer(palette = "Set1")
pbom <-
ggplot(df.heatmap |> filter(sample %in% s)) +
geom_raster(aes(x = x,y = y,fill = value)) +
theme_bw() +
theme(panel.grid = element_blank()) +
facet_grid2(split~.,switch = "y") +
xlab("") + ylab("") +
scale_fill_continuous_sequential(palette = "Reds 3") +
coord_cartesian(expand = 0) +
theme(strip.placement = "outside",
axis.ticks.y = element_blank(),
axis.text.y = element_blank(),
plot.margin = margin(t = 0,r = .2,b = .2,l = .2,
unit = "cm"),
panel.spacing.x = unit(0.6,"cm"),
axis.text.x = element_text(angle = 45,hjust = 1))
# merge
pmer <- aplot::plot_list(gglist = list(ptop,pbom),ncol = 1)
return(pmer)
}) -> plist
cowplot::plot_grid(plotlist = plist,nrow = 1)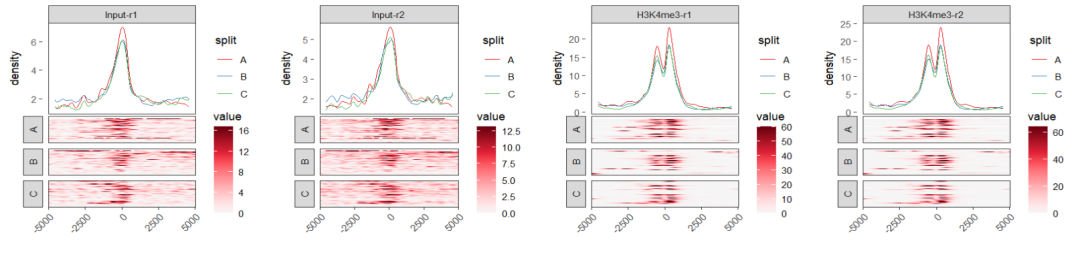
7结尾
路漫漫其修远兮,吾将上下而求索。
欢迎加入生信交流群。加我微信我也拉你进 微信群聊 老俊俊生信交流群 (微信交流群需收取 20 元入群费用,一旦交费,拒不退还!(防止骗子和便于管理)) 。QQ 群可免费加入, 记得进群按格式修改备注哦。
声明:文中观点不代表本站立场。本文传送门:https://eyangzhen.com/343570.html



