
1引言
相信大家见到的大部分环形图形有很多都是关于基因组的数据。ggcirclize 前面除了绘制一些 ggplot2 的基础图层和分面布局,对于基因组数据的支持也是必不可少的。构建一些 geom_trackgenomic… 相关的图层来可视化基因组的数据。
暂时写了:
geom_trackgenomicrect, geom_trackgenomicpoint, geom_trackgenomicline 和geom_trackgenomictile 这几个, 下面我们来看看一些效果。
2下载基因组信息
我们需要下载基因组的大小信息和染色体 cytoband 的信息,下面写一个简单的函数在 R 里下载,当然你也可以复制链接直接去网页下载:download_chromInfo <- function(genome = "hg19"){
url_chromsize <- paste0("https://hgdownload.soe.ucsc.edu/goldenPath/",
genome,
"/bigZips/",genome,".chrom.sizes")
download.file(url = url_chromsize,destfile = "./")
url_cytoband <- paste0("https://hgdownload.cse.ucsc.edu/goldenPath/",
genome,"/database/cytoBandIdeo.txt.gz")
download.file(url = url_cytoband,destfile = "./")
}
下载解压后就可以读进来了,我们同样写两个函数来过滤一些不想要的染色体:load_chrom_size <- function(genome = "hg19",chr = NULL){
chrom_size <- read.table(paste0("cytoband_chromsize/",genome,"_chrom_size.txt"))
# filter chromosome
if(is.null(chr)){
fil_chrom <- paste0("chr",c(1:22,"X","Y"))
}else{
fil_chrom <- chr
}
chrom_size <- subset(chrom_size,V1 %in% fil_chrom)
chrom_size <- chrom_size[match(fil_chrom,chrom_size$V1),]
return(chrom_size)
}
load_cytoband <- function(genome = "hg19",chr = NULL){
cytoBand <- read.delim(paste0("cytoband_chromsize/",genome,"_cytoBandIdeo.txt"),
header = F)
colnames(cytoBand) <- c("chr","start","end","band","stain")
# filter chromosome
if(is.null(chr)){
fil_chrom <- paste0("chr",c(1:22,"X","Y"))
}else{
fil_chrom <- chr
}
cytoBand <- subset(cytoBand,chr %in% fil_chrom)
return(cytoBand)
}
3geom_trackgenomictile
geom_trackgenomictile 来绘制矩形, cytoband 的信息就可以用来绘制染色体。
产生一些 bed 区间:library(circlize)
bed = generateRandomBed(nr = 300)
bed <- subset(bed,chr %in% c("chr1","chr2","chr3","chr4"))
bed$ymin <- sample(seq(0,1,0.1),nrow(bed),replace = T)
bed$ymax <- bed$ymin + 0.2
bed$group <- sample(LETTERS[1:4],nrow(bed),replace = T)
# check
head(bed)
# chr start end value1 ymin ymax group
# 1 chr1 114048 7101411 -0.2895206 0.1 0.3 D
# 2 chr1 8732752 11662832 -1.0575720 0.5 0.7 A
# 3 chr1 13705322 16977193 0.5415639 0.2 0.4 A
# 4 chr1 21827870 26896494 -0.7159598 0.8 1.0 B
# 5 chr1 28397265 30759921 -0.1669718 1.0 1.2 A
# 6 chr1 36176151 52241396 0.2176396 0.4 0.6 B
绘图:
chrom_data 参数用来加载染色体长度的信息,这个来分配当前 track 的每个 sector 的大小。
chrom_hg19 <- load_chrom_size(genome = "hg19")
ggcirclize(bed,
aes(end = 360,
chr = chr,gstart = start,gend = end)) +
geom_trackgenomicrect(aes(ymin = ymin,ymax = ymax),
chrom_data = chrom_hg19)
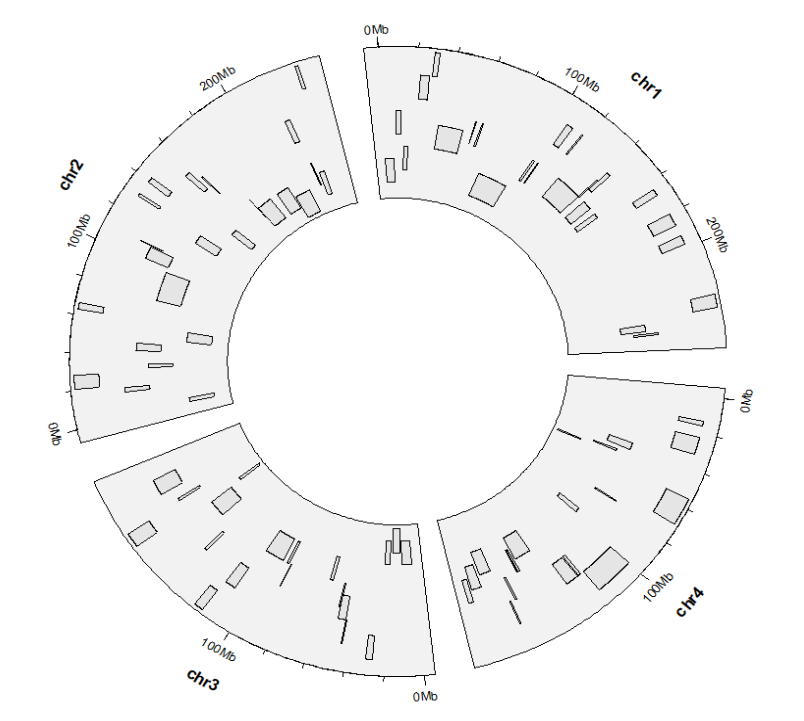

映射分组颜色:ggcirclize(bed,
aes(end = 360,
chr = chr,gstart = start,gend = end)) +
geom_trackgenomicrect(aes(ymin = ymin,ymax = ymax,fill = group),
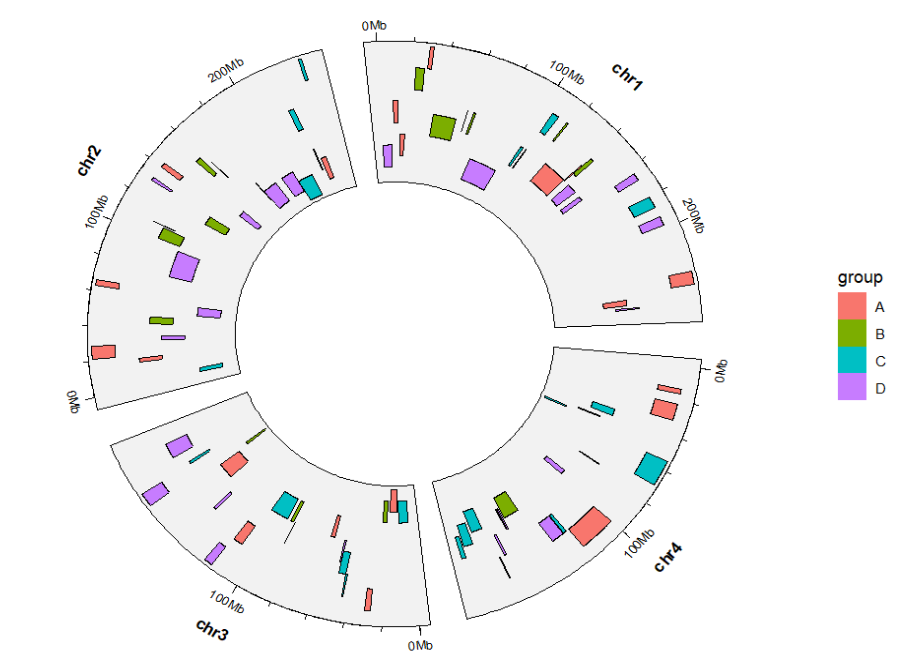
chrom_data = chrom_hg19)
加载染色体信息:cytoband_hg19 <- load_cytoband(genome = "hg19")
# check
head(cytoband_hg19,3)
# chr start end band stain
# 1 chr1 0 2300000 p36.33 gneg
# 2 chr1 2300000 5400000 p36.32 gpos25
# 3 chr1 5400000 7200000 p36.31 gneg
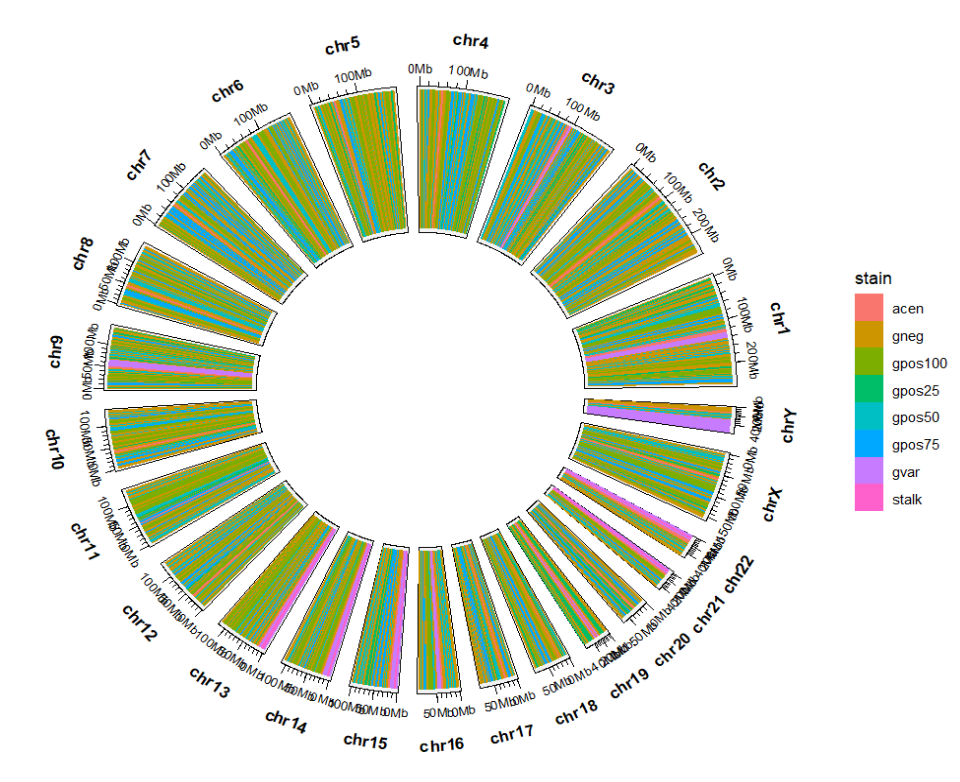
绘制染色体:p <-
ggcirclize(cytoband_hg19,
mg.t = 2,mg.b = 2,
aes(end = 360,
fill = stain,
chr = chr,gstart = start,gend = end)) +
geom_trackgenomicrect(color = NA,chrom_data = chrom_hg19)
p
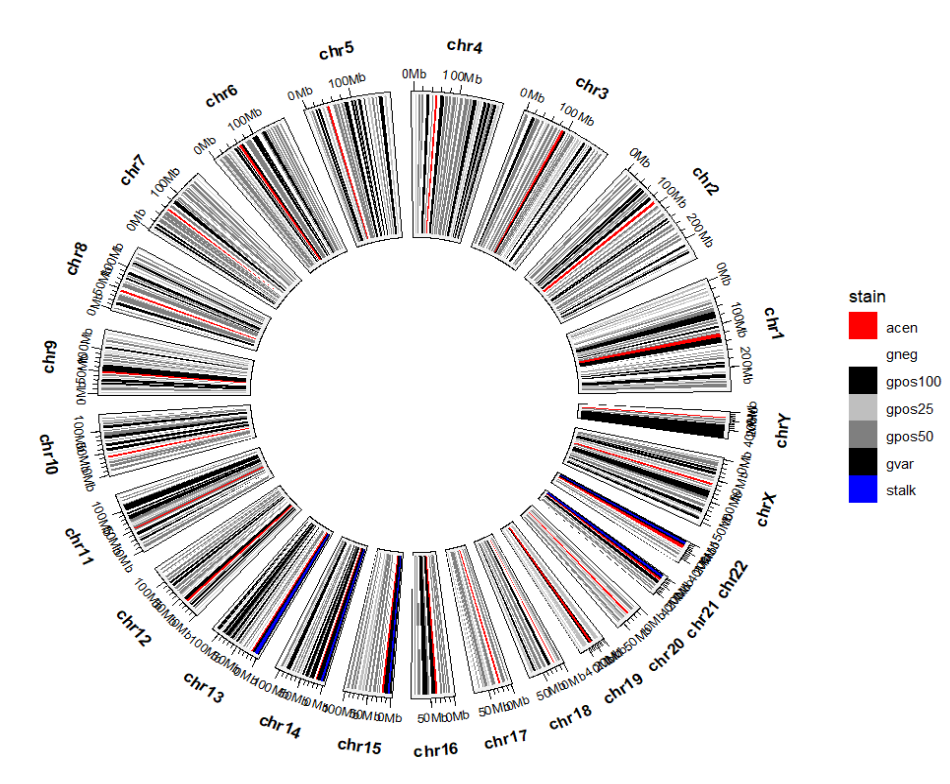
当然你可以换成常见的配色:p +
scale_fill_manual(values = c("gneg" = "white","gpos25" = "grey75","gpos50" = "grey50",
"gpos100" = "black","gvar" = "black","acen" = "red",
"stalk" = "blue"))
4geom_trackgenomicpoint
geom_trackgenomicpoint 绘制基因组范围里的散点图。
library(circlize)
bed = generateRandomBed(nr = 300)
chrom_hg19 <- load_chrom_size(genome = "hg19")
ggcirclize(bed,aes(end = 360,
chr = chr,gstart = start,gend = end,
value = value1)) +
geom_trackgenomicpoint(chrom_data = chrom_hg19)
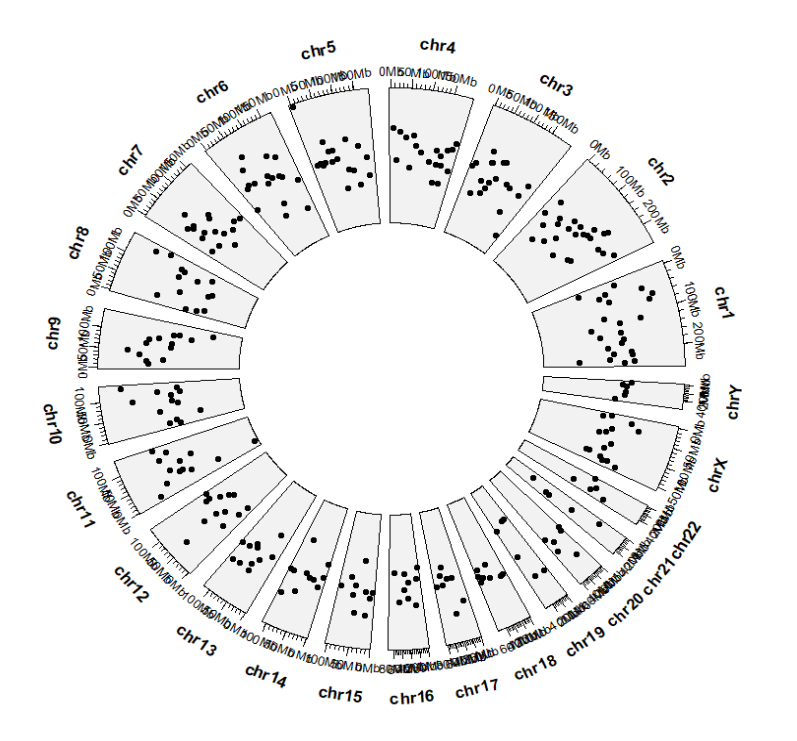
颜色映射:
ggcirclize(bed,aes(end = 360,
chr = chr,gstart = start,gend = end,
color = chr,
value = value1)) +
geom_trackgenomicpoint(chrom_data = chrom_hg19,
space = "fixed")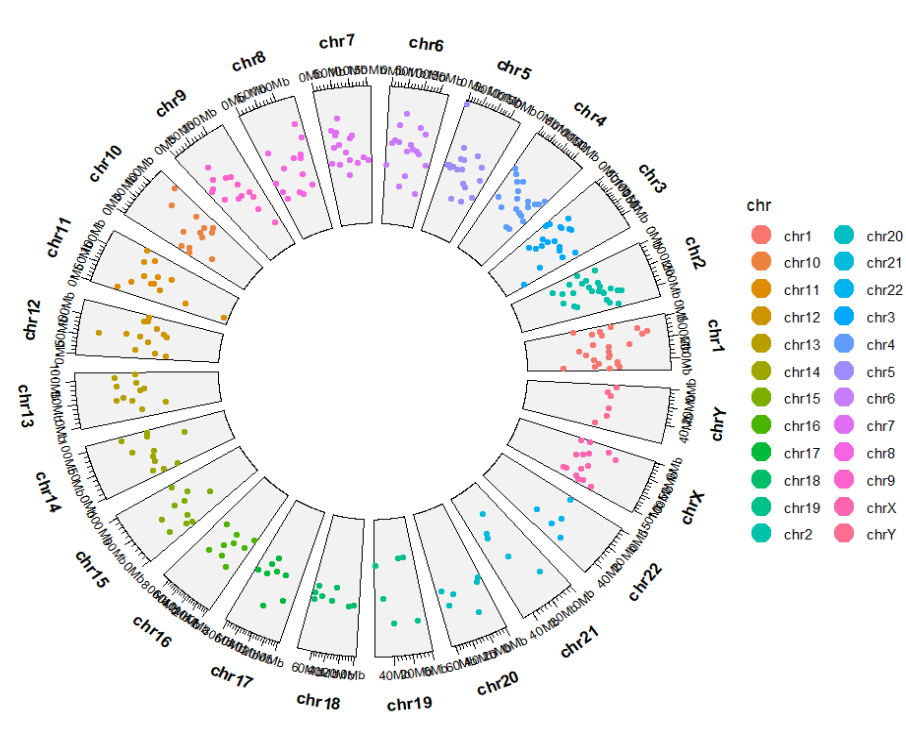
space = “fixed” 可以设置每个 sector 保持一样大小:bed <- subset(bed,chr %in% paste0("chr",1:6))
ggcirclize(bed,aes(end = 360,
chr = chr,gstart = start,gend = end,
color = chr,
value = value1)) +
geom_trackgenomicpoint(chrom_data = chrom_hg19,
space = "fixed")
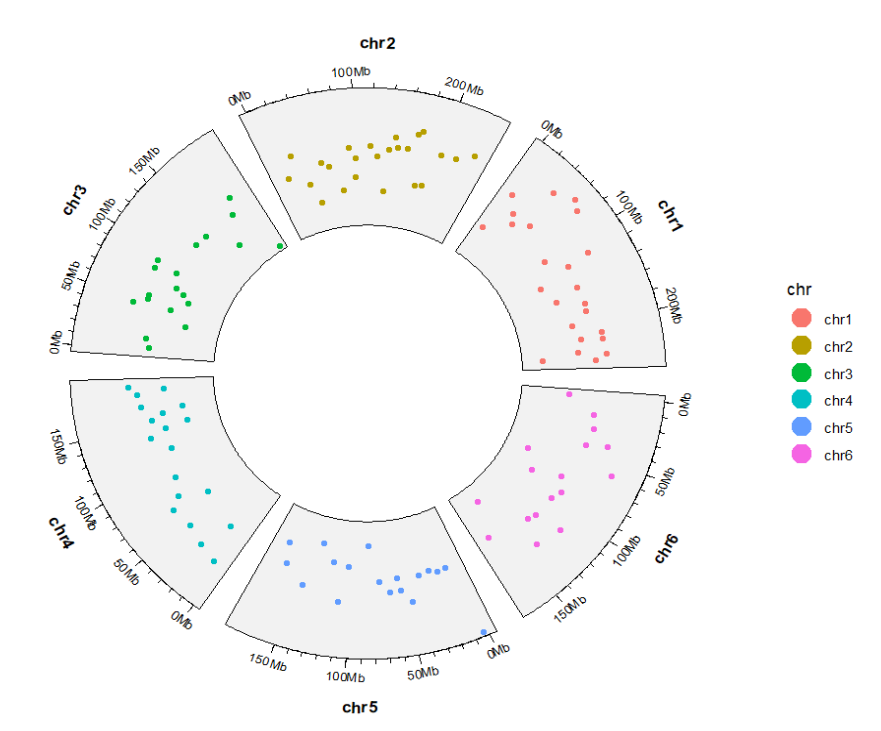
5geom_trackgenomicline
bed = generateRandomBed(nr = 300)
chrom_hg19 <- load_chrom_size(genome = "hg19")
ggcirclize(bed,aes(end = 360,color = chr,
chr = chr,gstart = start,gend = end,
value = value1)) +
geom_trackgenomicline(chrom_data = chrom_hg19)
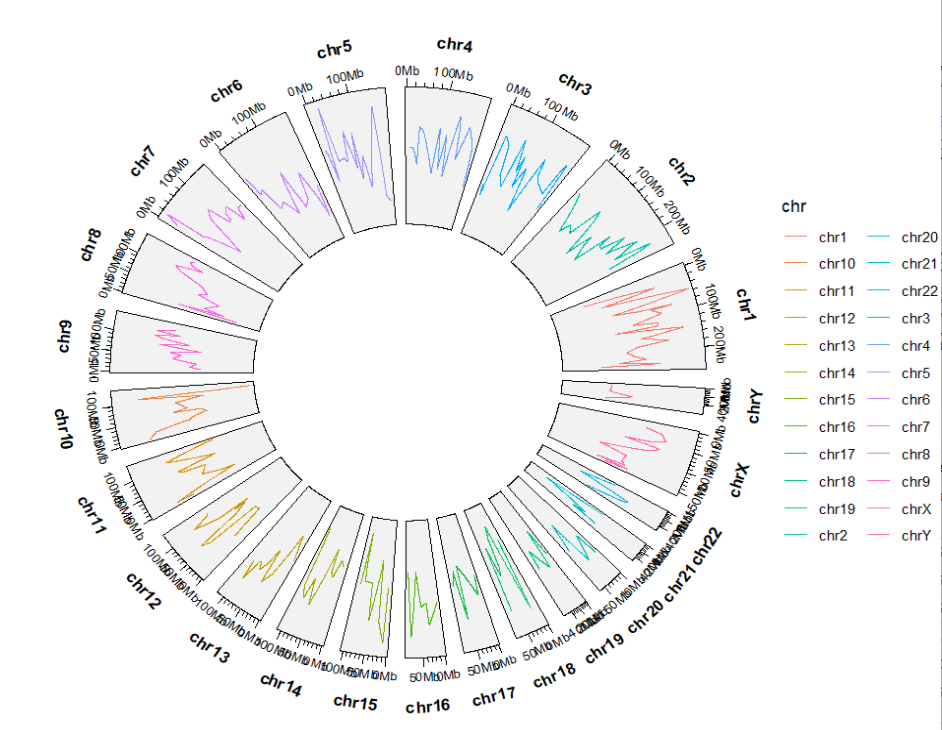
6geom_trackgenomictile
geom_trackgenomictile 用来绘制基因组热图。
热图基因组矩阵:bed = generateRandomBed(nr = 100, nc = 4)
# check
head(bed,3)
# chr start end value1 value2 value3 value4
# 1 chr1 4833203 25608564 0.4822349 0.2058859 0.01484909 -0.5005523
# 2 chr1 48610396 76802798 0.7022751 0.4851126 -1.38951435 0.6165077
# 3 chr1 79055730 87730120 -0.3187775 0.4399068 0.51394522 0.2435110
定义一个函数将上面宽格式的转换为长格式的:# define func
bedMatTolong <- function(bed.mat = NULL,retain.var = NULL){
bed.mat <- bed.mat[order(bed.mat$chr,bed.mat$start),]
x <- as.numeric(unlist(lapply(table(bed.mat$chr), function(x) 1:x)))
bed.mat$x <- x
if(is.null(retain.var)){
id.vars <- c("chr","start","end","x")
}else{
id.vars <- c("chr","start","end","x",retain.var)
}
bed_long <- reshape2::melt(bed.mat,id.vars = id.vars)
bed_long$y <- as.numeric(bed_long$variable)
return(bed_long)
}
bed_long <- bedMatTolong(bed)
head(bed_long,3)
# chr start end x variable value y
# 1 chr1 4833203 25608564 1 value1 0.4822349 1
# 2 chr1 48610396 76802798 2 value1 0.7022751 1
# 3 chr1 79055730 87730120 3 value1 -0.3187775 1
绘图:ggcirclize(bed_long,aes(end = 360,r0 = 0.5,r1 = 0.8,
chr = chr,gstart = start,gend = end)) +
geom_trackgenomictile(aes(x = x,y = y,fill = value),
chrom_data = chrom_hg19,strip.label = T) +
scale_fill_gradient2(low = "green",mid = "white",high = "red",midpoint = 0)
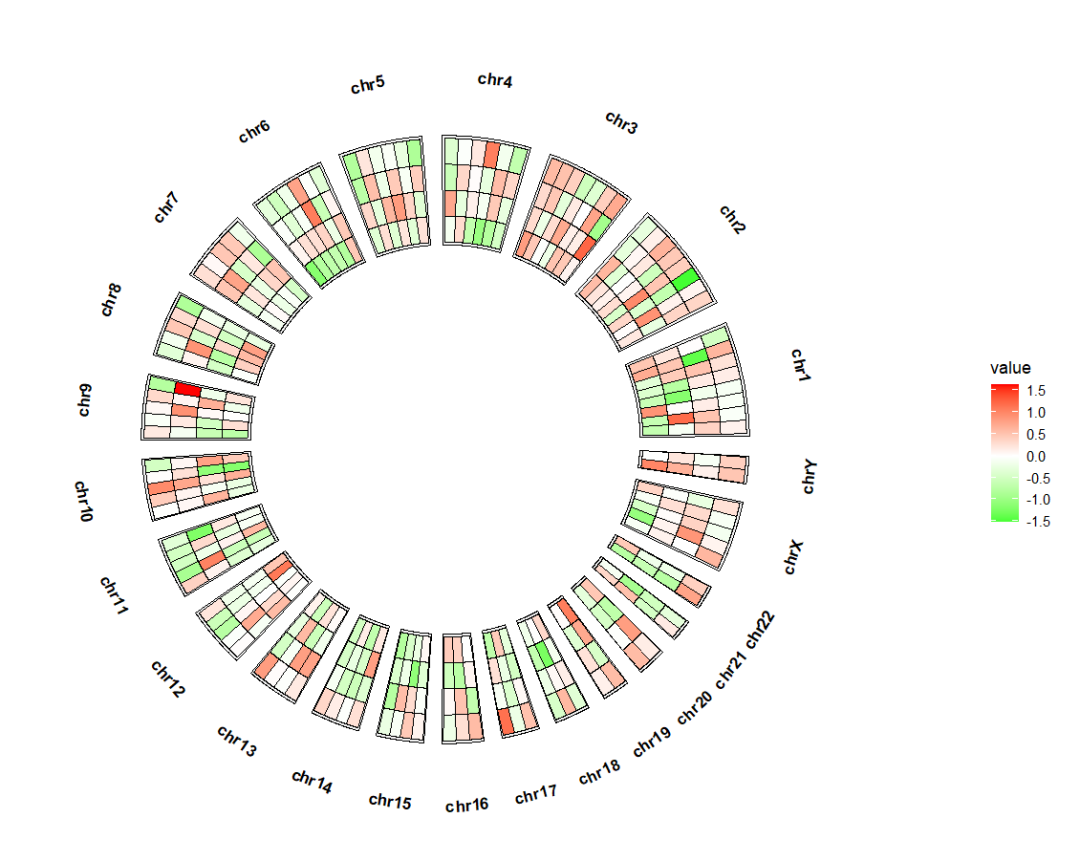
add_link 参数添加原始基因组位置与热图位置的连接,参考 circlize 的基因组热图。
ggcirclize(bed_long,aes(end = 360,r0 = 0.5,r1 = 0.7,
chr = chr,gstart = start,gend = end)) +
geom_trackgenomictile(aes(x = x,y = y,fill = value),
chrom_data = chrom_hg19,strip.label = T,
add_link = T,
link_pos = "top") +
scale_fill_gradient2(low = "green",mid = "white",high = "red",midpoint = 0)
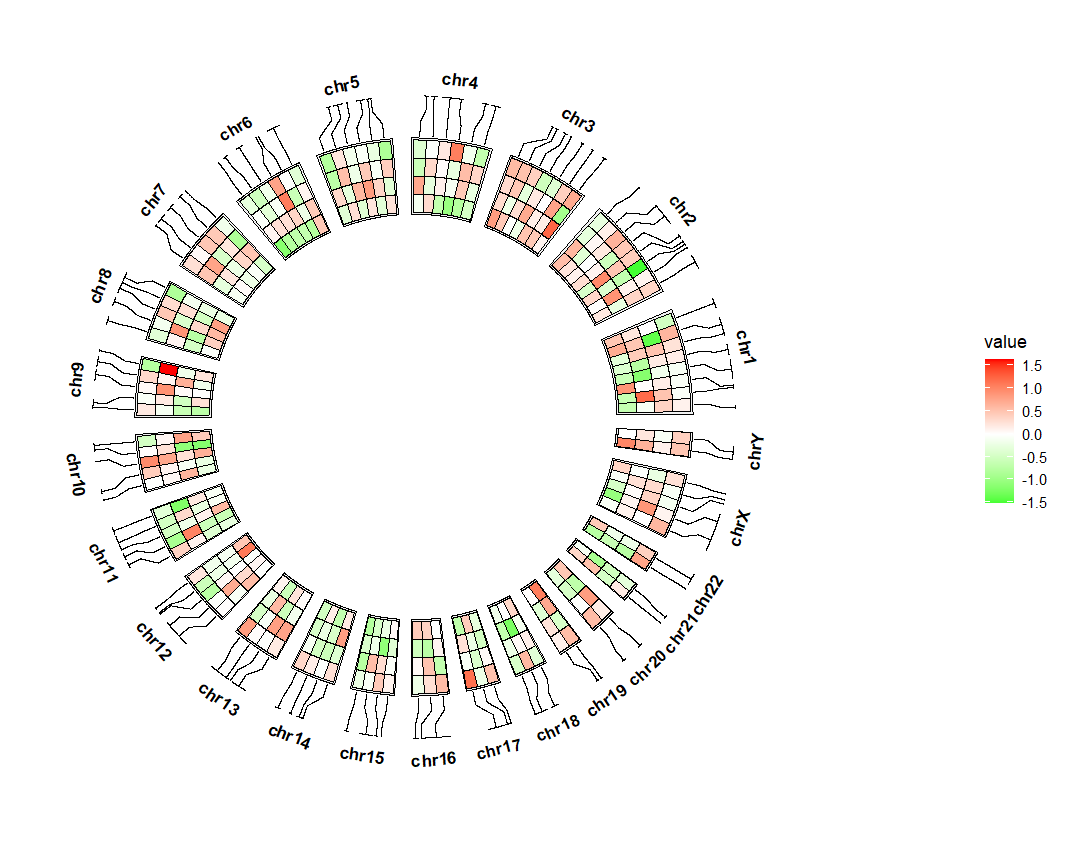
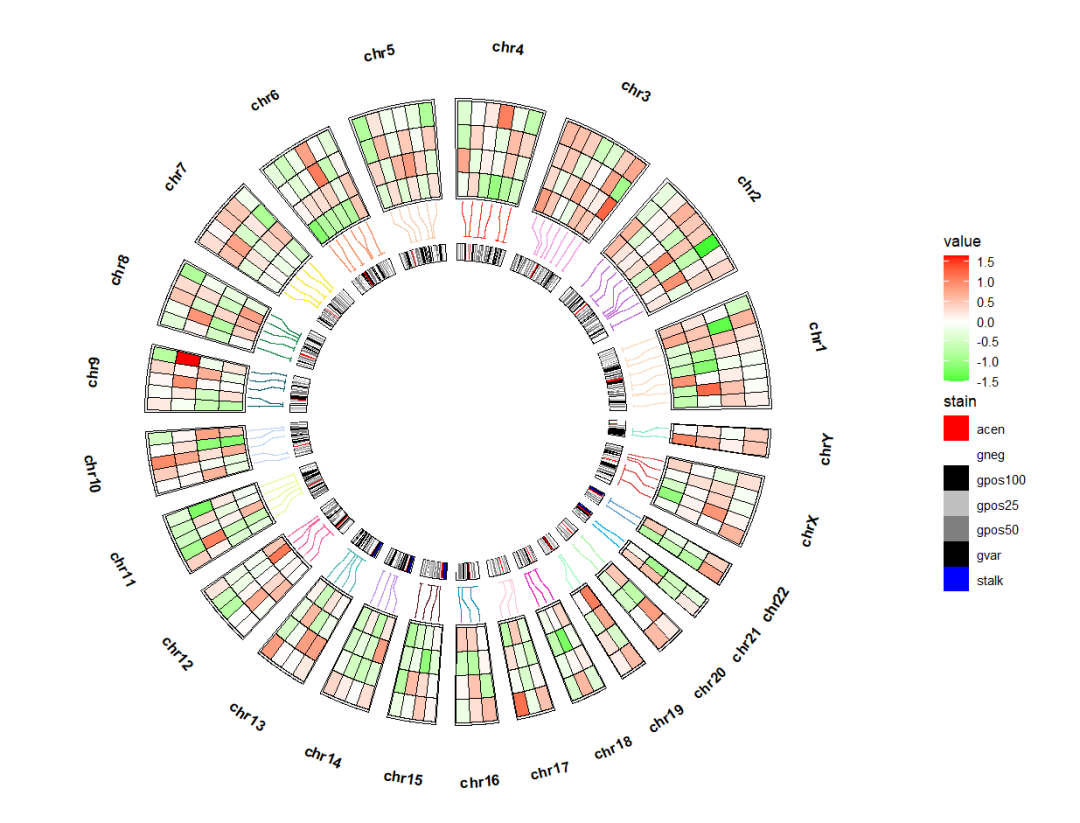
link_pos 指定位置,比如放置下面, link_col 设置连接线的颜色:ggcirclize(bed_long,aes(end = 360,r0 = 0.5,r1 = 0.7,
chr = chr,gstart = start,gend = end)) +
geom_trackgenomictile(aes(x = x,y = y,fill = value),
chrom_data = chrom_hg19,strip.label = T,
add_link = T,
link_pos = "bottom",
link_col = circlize::rand_color(24)) +
scale_fill_gradient2(low = "green",mid = "white",high = "red",midpoint = 0)
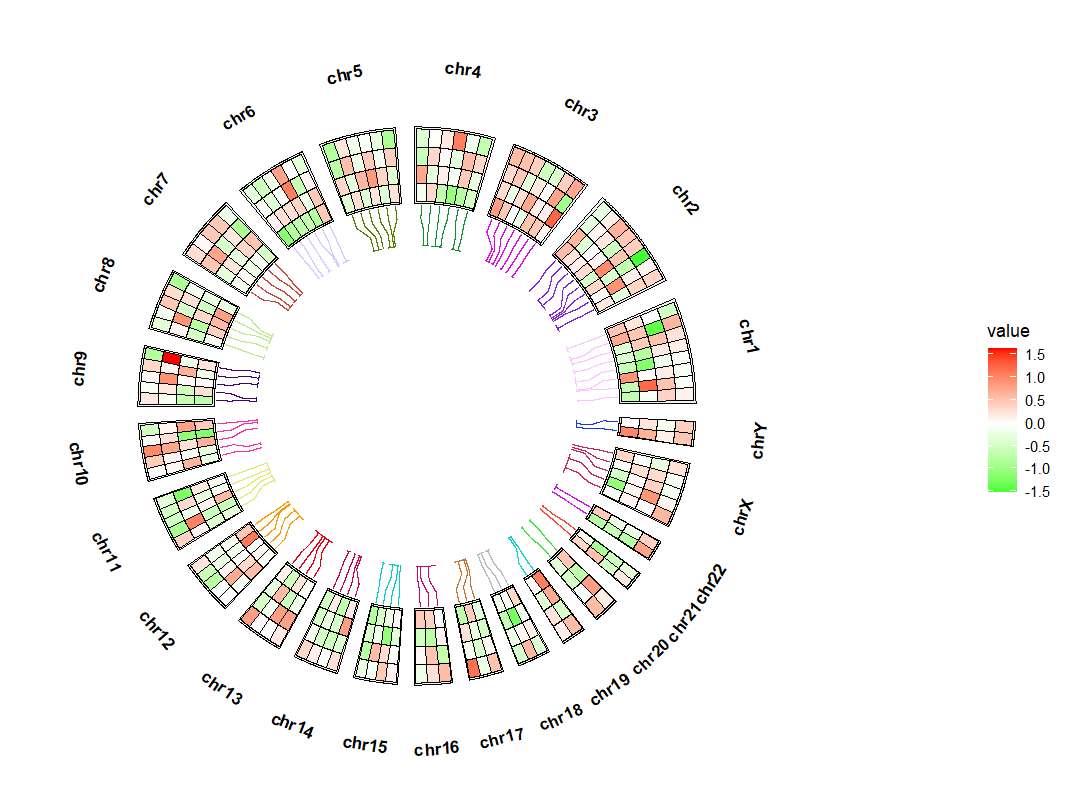
link_r 控制连接线的长短:ggcirclize(bed_long,aes(end = 360,r0 = 0.5,r1 = 0.7,
chr = chr,gstart = start,gend = end)) +
geom_trackgenomictile(aes(x = x,y = y,fill = value),
chrom_data = chrom_hg19,strip.label = T,
add_link = T,
link_pos = "bottom",
link_col = circlize::rand_color(24),
link_r = 0.3) +
scale_fill_gradient2(low = "green",mid = "white",high = "red",midpoint = 0)
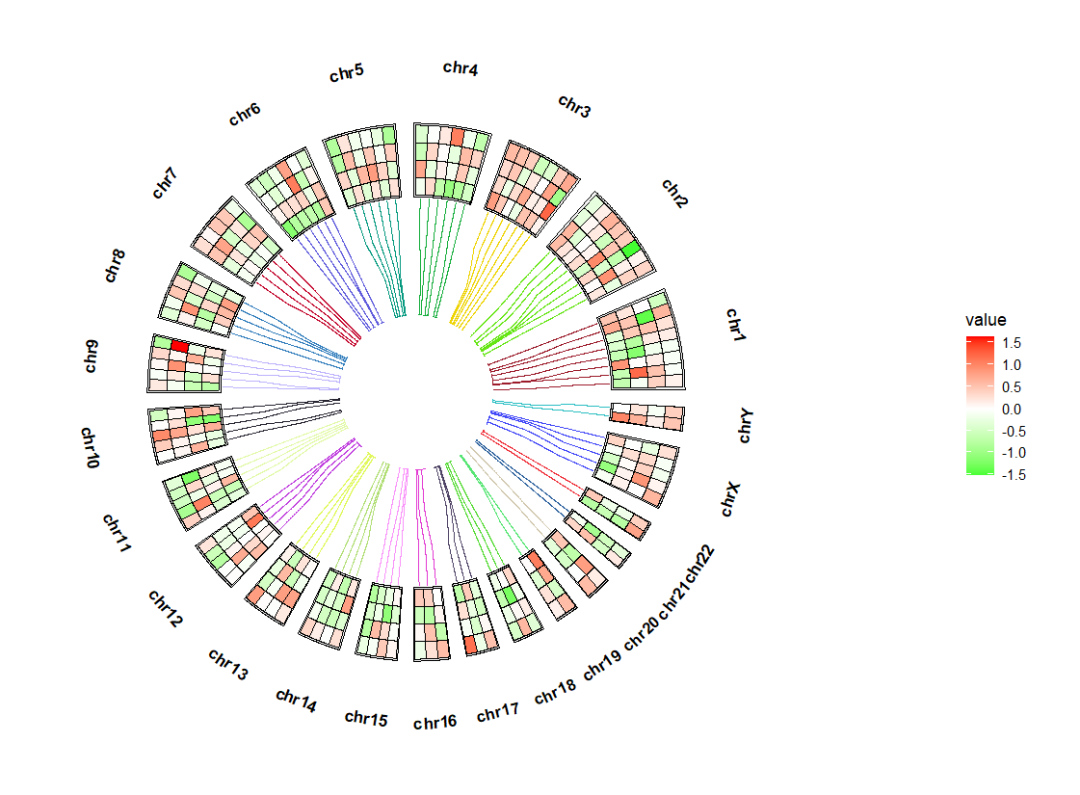
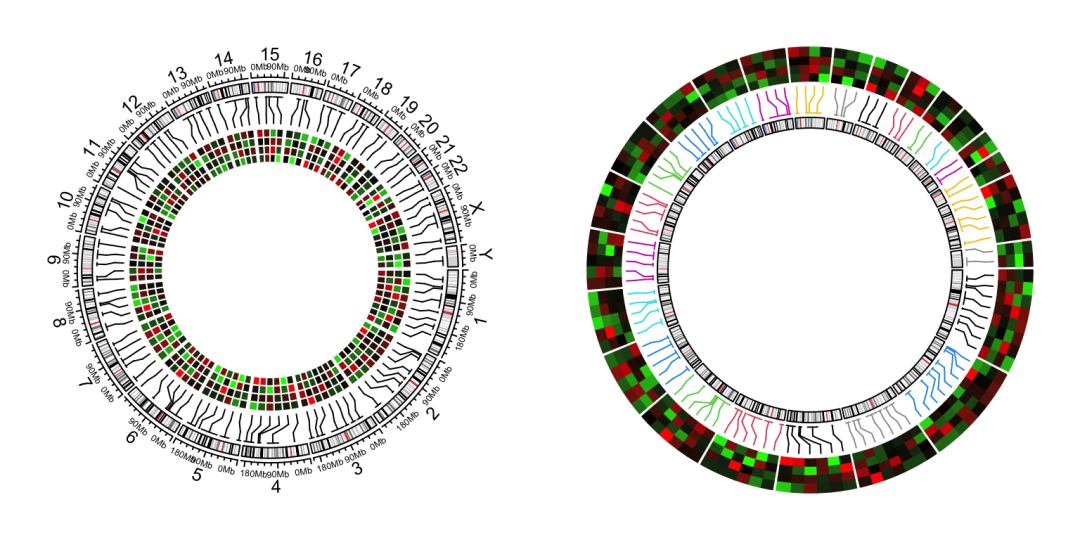
我们把染色体也画出来:library(ggnewscale)
ggcirclize(bed_long,aes(end = 360,r0 = 0.4,r1 = 0.68,
chr = chr,gstart = start,gend = end)) +
geom_trackgenomictile(aes(x = x,y = y,fill = value),
chrom_data = chrom_hg19,strip.label = F,
add_link = T,
link_pos = "top",
link_col = circlize::rand_color(24)) +
scale_fill_gradient2(low = "green",mid = "white",high = "red",midpoint = 0) +
# ============================================================================
new_scale_fill() +
geom_trackgenomicrect(data = cytoband_hg19,
aes(r0 = 0.8,r1 = 0.85,fill = stain),
color = NA,chrom_data = chrom_hg19) +
scale_fill_manual(values = c("gneg" = "white","gpos25" = "grey75","gpos50" = "grey50",
"gpos100" = "black","gvar" = "black","acen" = "red",
"stalk" = "blue"))
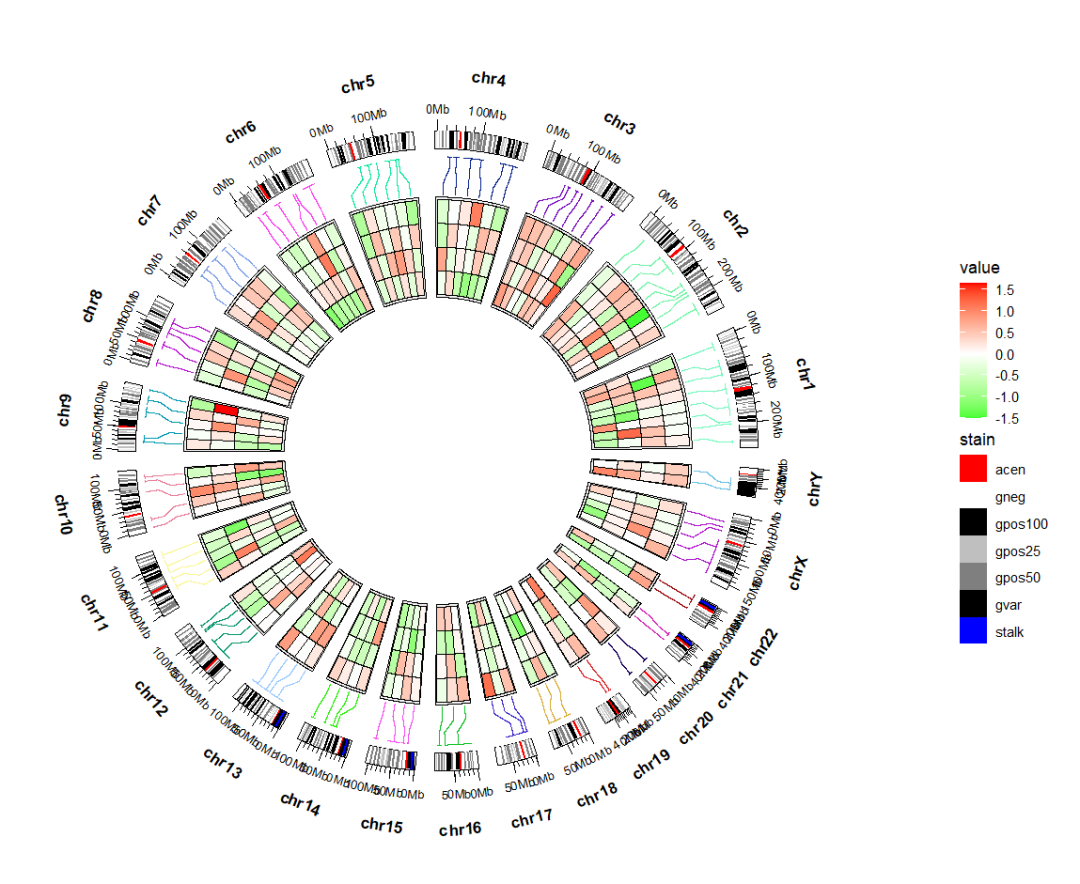
换个位置:ggcirclize(bed_long,aes(end = 360,r0 = 0.6,r1 = 0.9,
chr = chr,gstart = start,gend = end)) +
geom_trackgenomictile(aes(x = x,y = y,fill = value),
chrom_data = chrom_hg19,strip.label = T,
add_link = T,
link_pos = "bottom",
link_col = circlize::rand_color(24)) +
scale_fill_gradient2(low = "green",mid = "white",high = "red",midpoint = 0) +
# ============================================================================
new_scale_fill() +
geom_trackgenomicrect(data = cytoband_hg19,
strip.label = F,
add.xaxis = F,
aes(r0 = 0.43,r1 = 0.48,fill = stain),
color = NA,chrom_data = chrom_hg19) +
scale_fill_manual(values = c("gneg" = "white","gpos25" = "grey75","gpos50" = "grey50",
"gpos100" = "black","gvar" = "black","acen" = "red",
"stalk" = "blue"))
7结尾
路漫漫其修远兮,吾将上下而求索。
欢迎加入生信交流群。加我微信我也拉你进 微信群聊 老俊俊生信交流群 (微信交流群需收取 20 元入群费用,一旦交费,拒不退还!(防止骗子和便于管理)) 。QQ 群可免费加入, 记得进群按格式修改备注哦。
声明:文中观点不代表本站立场。本文传送门:https://eyangzhen.com/384092.html



