
论文
Pangenome analysis reveals genomic variations associated with domestication traits in broomcorn millet
论文中提供大部分图的原始作图数据,我们可以试着用论文中提供的原始数据来复现一下论文中的图
今天的推文来复现一下论文中的figure2g
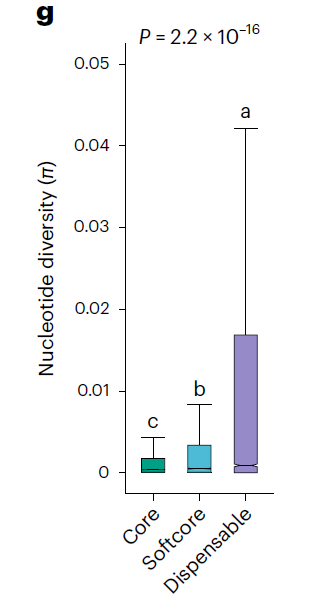

今天的推文主要是学习这里做差异检验的方法,figure2的图注里写到
Significance was tested using a Kruskal–Wallis test; multiple comparisons were analyzed using a Nemenyi test. The different lowercase letters above the box plots represent significant differences (P ≤ 0.05).
论文中提供的示例数据截图
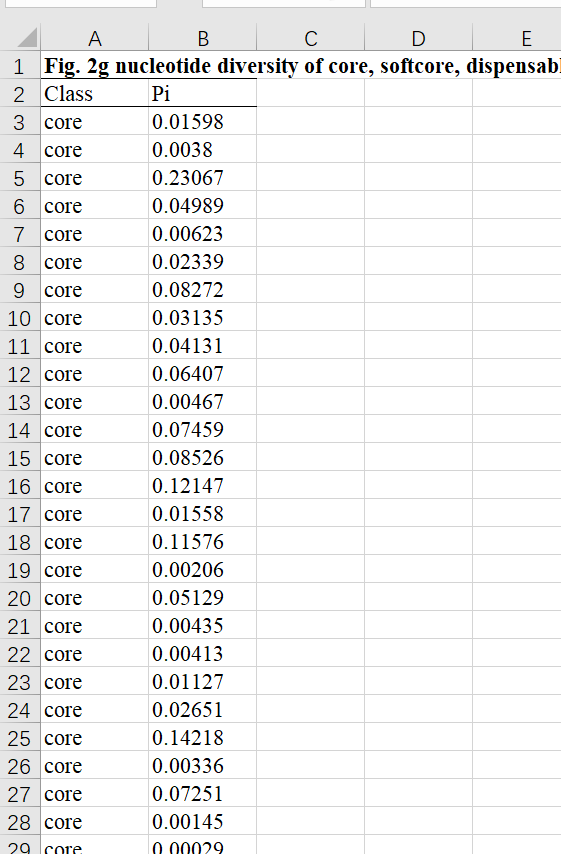
读取数据
library(readxl)
fig2g.dat<-read_excel("data/20231201/41588_2023_1571_MOESM5_ESM.xlsx",
sheet = "Fig. 2g",
skip = 1)
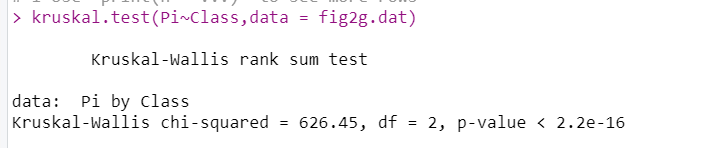
fig2g.datKruskal–Wallis test
kruskal.test(Pi~Class,data = fig2g.dat)
多重比较 a Nemenyi test
这里需要给分组变量添加因子水平
fig2g.dat %>%
mutate(Class=factor(Class)) -> fig2g.dat
#install.packages("PMCMRplus")
PMCMRplus::kwAllPairsNemenyiTest(Pi~Class,data = fig2g.dat)
PMCMRplus::kwAllPairsNemenyiTest(Pi~Class,data = fig2g.dat,dist="Chisquare")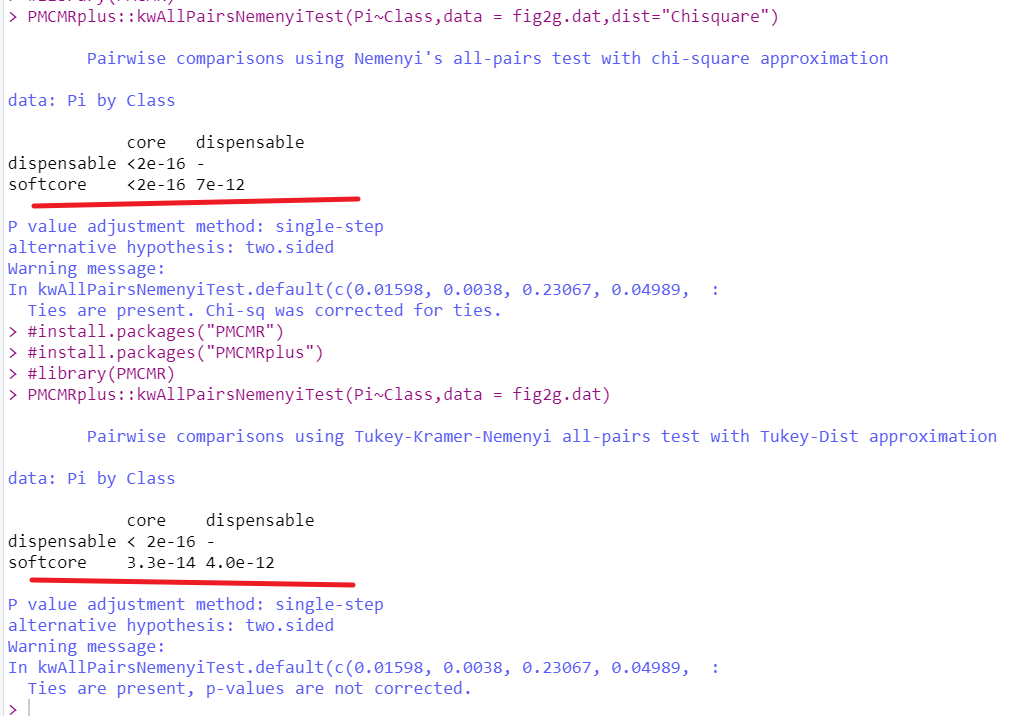
作图代码
直接用论文中提供的数据做出的效果如下
ggplot(data=fig2g.dat,aes(x=Class,y=Pi))+
geom_boxplot()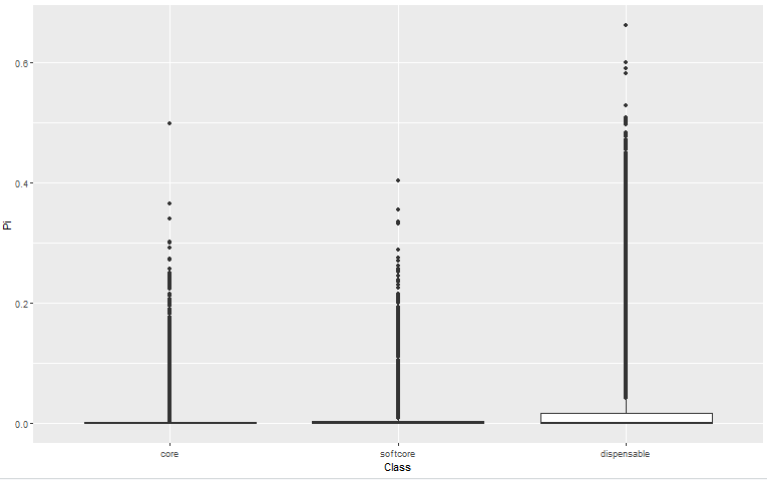
把y轴的范围限制到0到0.05,去掉离群值
ggplot(data=fig2g.dat,aes(x=Class,y=Pi))+
geom_boxplot(outlier.alpha = 0)+
scale_y_continuous(limits = c(0,0.05))+
theme_bw()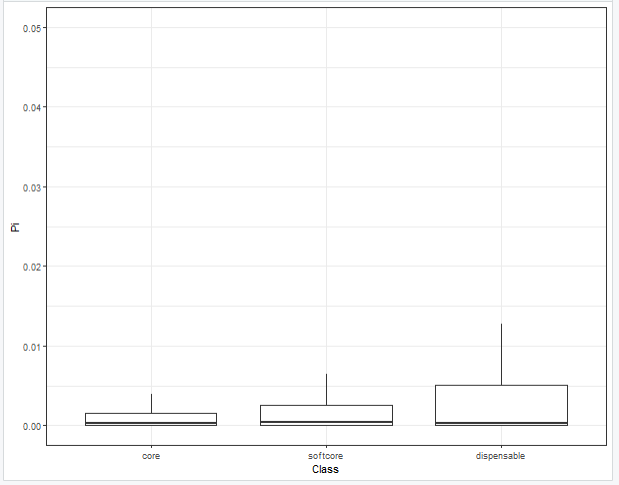
这个和论文中最终呈现的图还是不太一致,暂时没太想明白论文中是怎们处理这部分数据用于作图的,我个人感觉把核苷酸多样性取log10再作图看起来会美观一点,不知道这样处理是否合适
ggplot(data=fig2g.dat,aes(x=Class,y=Pi))+
stat_boxplot(geom = "errorbar",
width=0.2)+
geom_boxplot(aes(fill=Class),notch=TRUE,
notchwidth = 0.1,
outlier.alpha = 0,
width=0.3)+
theme_bw(base_size = 20)+
theme(panel.grid = element_blank(),
legend.position = "none",
axis.text.x = element_text(angle=60,vjust=1,hjust=1))+
labs(x=NULL,y="Nucleotide diversity (u03c0)")+
scale_y_log10(breaks=c(0.00001,0.0001,0.001,0.01,0.1),
labels = scales::trans_format("log10", scales::math_format(10^.x)))+
scale_fill_manual(values = c("#00a087","#4dbbd5","#968bc8"))+
geom_text(data=data.frame(x=c(1,2,3),y=c(0.2,0.6,0.99),label=c("c","b","a")),
aes(x=x,y=y,label=label))+
annotate(geom = "text",x=2,y=1.5,label="p=2.22x10-16")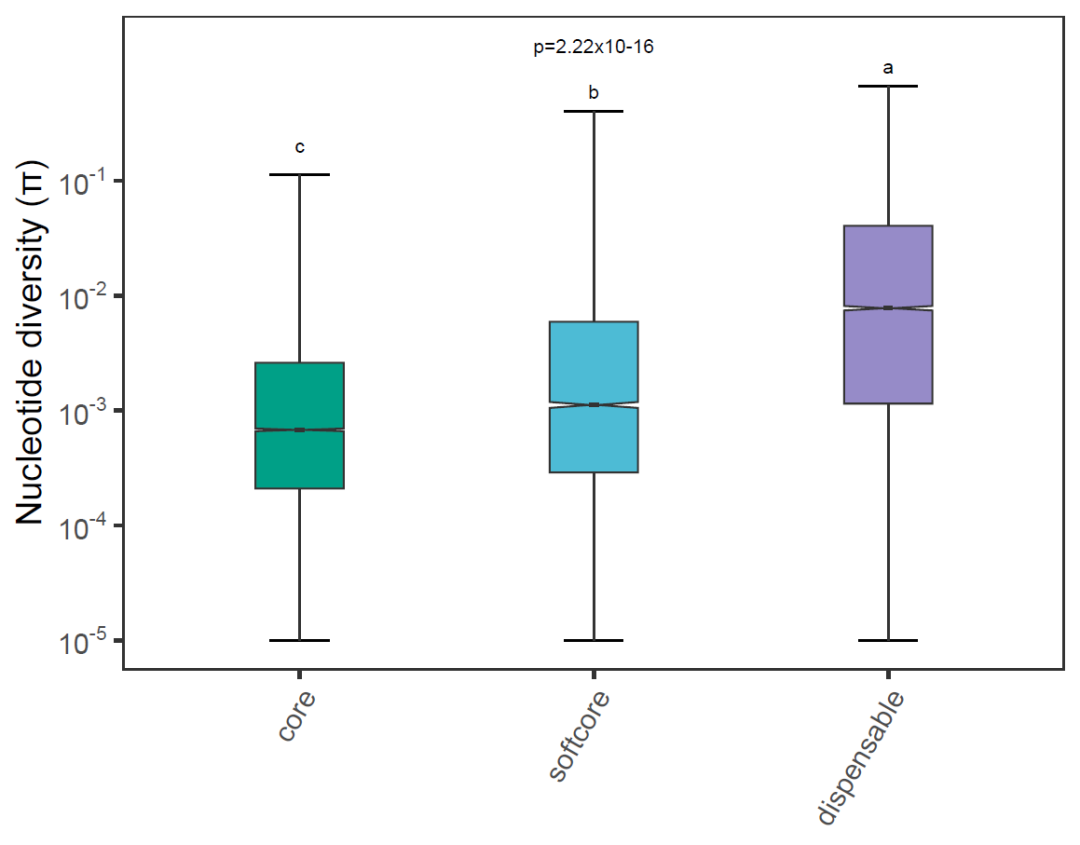
示例数据可以到论文中下载,或者给推文打赏1元获取我整理的示例数据和代码
欢迎大家关注我的公众号
小明的数据分析笔记本
声明:文中观点不代表本站立场。本文传送门:https://eyangzhen.com/385428.html



