
使用到的R包 doubletrouble,这个R包 对应的论文
Doubletrouble: Identification and Classification of Duplicated Genes
可以用来鉴定基因组中的重复基因
这个论文里还提到一个R包syntenet
syntenet: an R/Bioconductor package for the inference and analysis of synteny networks
推断和分析同线性网络,这个推断的作用:鉴定某个类群特异的 gene cluster,这些特异的cluster可能对这个类群重要的表型特征有影响
highlight taxon-specific gene clusters that likely contributed to the evolution of important traits, and microsynteny-based phylogenies can help resolve phylogenetic relationships under debate.
还提到一个R包 MSA2dist 主要是读取处理DNA和蛋白序列 帮助文档
今天推文的主要内容是已经有cds序列,计算kaks值。今天推文里用到的cds序列来源于论文
Beginner’s Guide on the Use of PAML to Detect Positive Selection
这个论文有时间要看看,介绍的是PAML这个软件计算Dn/Ds
github主页
第一步是读取cds序列
dna<-Biostrings::readDNAStringSet("data1_unaln.fasta")
生成序列id的两两组合gene_pairs<-as.data.frame(t(combn(names(dna),2)))
计算kaks值
cds_list<-list(kakspra=dna)
gene.pairs_list<-list(kakspra=gene_pairs)
kaks <- pairs2kaks(gene.pairs_list, cds_list)
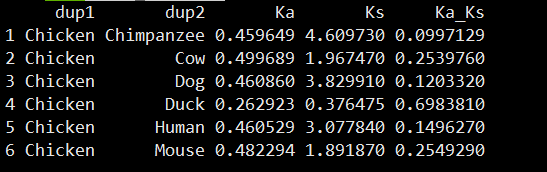
head(kaks$kakspra)
这里试了一下序列id里如果有下划线还不行,需要把下滑线去掉

声明:文中观点不代表本站立场。本文传送门:https://eyangzhen.com/413310.html



