
1引言
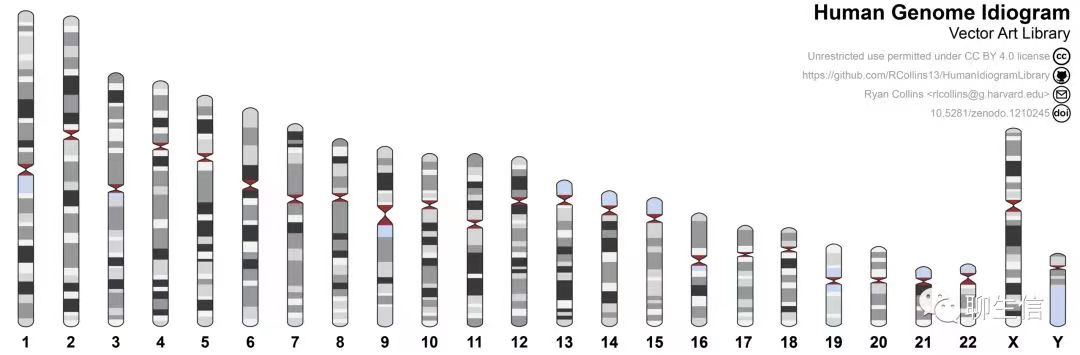

上面这张图想必大家都见到过, 但是你有没有想过自己怎样去画一个这样的呢? 目前是有一些这样的包的,比如 Gviz, Idiogram 等。你知道这个图的意思是什么吗? 我也不知道, 于是去搜索了资料,做了一些功课。上面代表了 24 条染色体结构, 染色体上的颜色深度代表基因的密度,中间红色的地方代表着丝粒的位置。 既然知道了,是不是自己就可以手动来绘制这样的图了呢?
输入数据:
首先你得知道我们需要哪些数据来绘图:
- 首先你需要每条染色体的大小
- 还需要每条染色体着丝粒的位置
- 然后你需要计算基因的密度
- 最后绘图即可
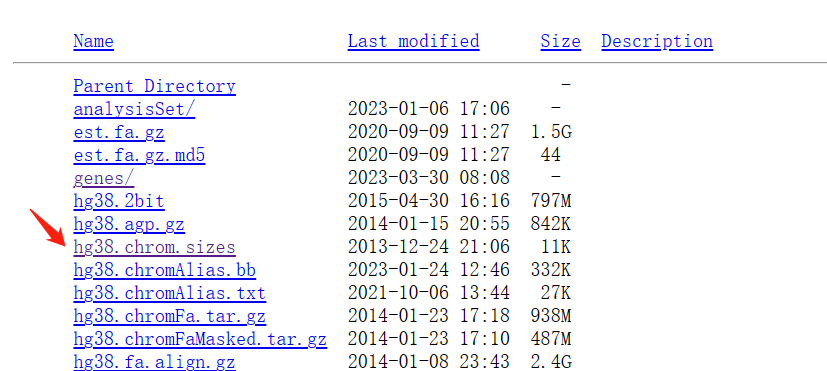
数据从哪下呢,染色体大小你可以在 UCSC 官网下载,选择对应的基因组,找到 .chrom.sizes 结尾的即可:
着丝粒 位置的数据可以参考推文 获取人类染色体长度及着丝粒(Centromere )和端粒(Telomere)位置, 接下来本文手把手教你绘制过程, 为了方便,我已包装了一些函数在 BioSeqUtils 里,直接调用即可。
2安装
重新安装获取新功能:
# install.packages("devtools")
devtools::install_github("junjunlab/BioSeqUtils")
library(BioSeqUtils)3正文
我已经整理了 “hg19”, “hg38”, “mm10”, “mm39” 这些基因组的 染色体长度和着丝粒 的数据,直接加载即可。_ce_start_ 和 ce_end 是该染色体着丝粒的起始和终止位置, start 和 end 是染色体的起始和终止位置。
library(BioSeqUtils)
library(dplyr)
load("all_karyotype.rda")
# check
head(all_karyotype,3)
# chr ce_start ce_end start end type
# 1 chr1 121535434 124535434 0 249250621 hg19
# 2 chr2 92326171 95326171 0 243199373 hg19
# 3 chr3 90504854 93504854 0 198022430 hg19
unique(all_karyotype$type)
# [1] "hg19" "hg38" "mm10" "mm39"你也可以使用 getChromSize 函数获取你自己基因组的长度:
myobj <- loadGenomeGTF(gtfPath = "hg38.ncbiRefSeq.gtf.gz",
genomePath = "hg38.fa")
## GenomeGTF object for Extracting sequences.
## GTF file is loaded.
## genome file is loaded.
## representTrans file is NULL.
## intron slot is NULL.
# get chromosome size
chromSize <- getChromSize(object = myobj)
head(chromSize)
# chr start end
# 1 chr1 0 248956422
# 2 chr10 0 133797422
# 3 chr11 0 135086622
# 4 chr11_KI270721v1_random 0 100316
# 5 chr12 0 133275309
# 6 chr13 0 114364328你可以使用 prepareKaryotype 函数来整理成开头数据的格式,只需要提供染色体大小和着丝粒的位置文件, 如果没有丝粒的位置文件,则位置设置为 0。
prepareKaryotype(chromSize = chromSize,centromere = NULL) %>%
head()
# chr start end ce_start ce_end
# 1 chr1 0 248956422 0 0
# 2 chr10 0 133797422 0 0
# 3 chr11 0 135086622 0 0
# 4 chr11_KI270721v1_random 0 100316 0 0
# 5 chr12 0 133275309 0 0
# 6 chr13 0 114364328 0 0数据得保证你的染色体名称和 gtf 注释文件是一样的,要么都是 chr 开头,要么都没有。使用getGeneDensiy 函数来计算染色体上基因的参数,需要上面准备的 karyotype 文件和 注释文件, feature参数指定计算的指标,默认是 transcript,如果你的注释文件 type 列有 gene,你也可以指定它, step 参数指定将染色体分割的窗口大小。
gtf_file <- myobj@gtf %>% as.data.frame()
# check
head(gtf_file[1:3,1:10])
# seqnames start end width strand source type score phase gene_id
# 1 chrM 15956 16023 68 - ncbiRefSeq.2022-10-28 transcript NA NA TRNP
# 2 chrM 15956 16023 68 - ncbiRefSeq.2022-10-28 exon NA NA TRNP
# 3 chrM 15888 15953 66 + ncbiRefSeq.2022-10-28 transcript NA NA TRNT
karyotype.df <- all_karyotype %>% dplyr::filter(type == "hg38")
density.df <- getGeneDensiy(karyotype = karyotype.df,gtf = gtf_file,
feature = "transcript",
step = 10*10^6)
# check
head(density.df,3)
# chr start end type numGenes density
# 1 chr1 1e+00 1e+07 hg38 1269 0.006625698
# 2 chr1 1e+07 2e+07 hg38 1007 0.005257744
# 3 chr1 2e+07 3e+07 hg38 1347 0.007032951calcuCentromereData 计算着丝粒坐标,用来绘制中间三角形的区域。
# centromere data
centromere.triangle <- calcuCentromereData(karyotype.df)
# check
head(centromere.triangle,3)
# chr x y
# 1 chr1 121700000 0.0
# 2 chr1 121700000 1.0
# 3 chr1 123400000 0.5drawChromosome 函数来绘图, 绘制所有染色体结构:
# plot
drawChromosome(karyotype_file = karyotype.df,
centromereTri_file = centromere.triangle,
density_file = density.df,
chromosomes = karyotype.df$chr,
facet_params = list(scales = "fixed"))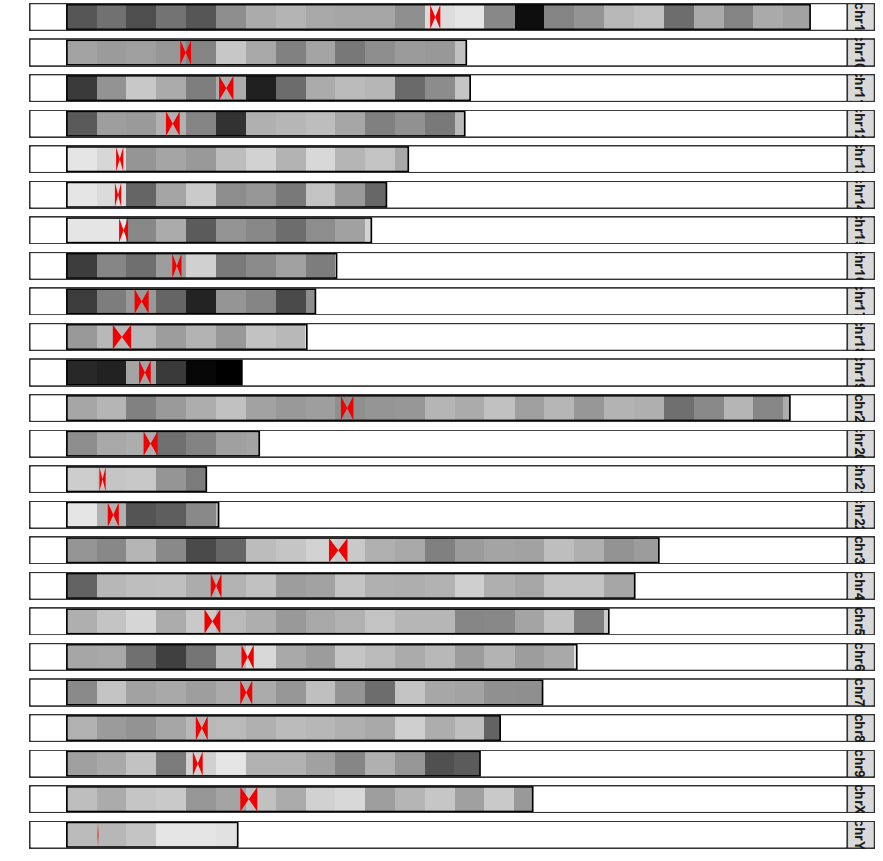
选择部分染色体绘制:
drawChromosome(karyotype_file = karyotype.df,
centromereTri_file = centromere.triangle,
density_file = density.df,
chromosomes = c("chr1","chr2","chr3"),
facet_params = list(strip_position = "left"))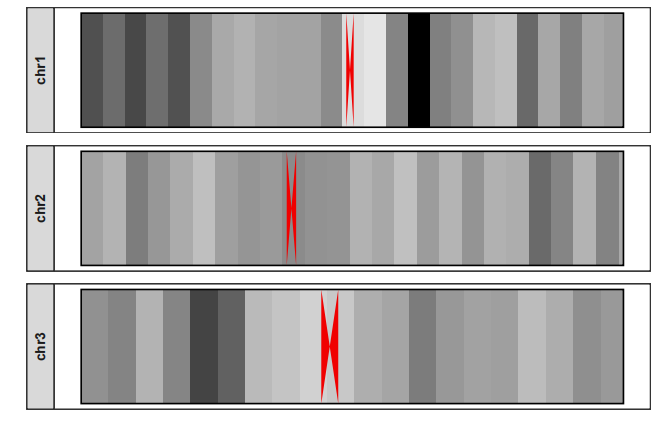
使用 zoom_region 来标注某个区域:
drawChromosome(karyotype_file = karyotype.df,
centromereTri_file = centromere.triangle,
density_file = density.df,
chromosomes = c("chr1","chr2","chr3"),
facet_params = list(strip_position = "left"),
zoom_region = c(10^8,10^8+300000))
放置在下面:
drawChromosome(karyotype_file = karyotype.df,
centromereTri_file = centromere.triangle,
density_file = density.df,
chromosomes = c("chr1","chr2","chr3"),
facet_params = list(strip_position = "left"),
zoom_region = c(10^8,10^8+300000),
zoom_pos = "bottom")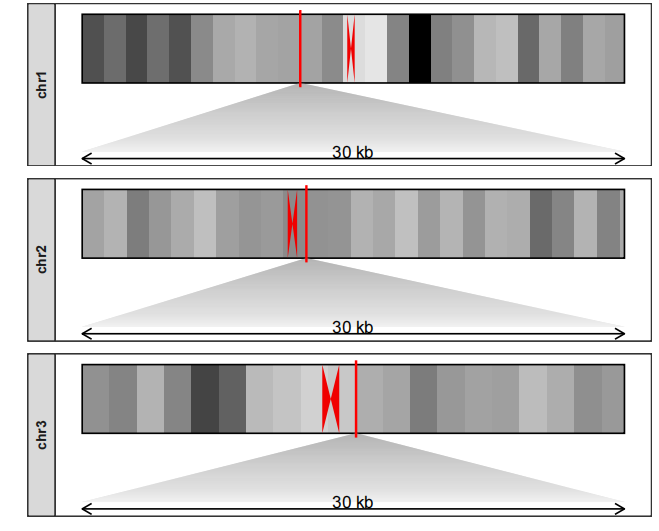
对不同染色体不同区域标注, zoom_relheight 设置相对高度:
drawChromosome(karyotype_file = karyotype.df,
centromereTri_file = centromere.triangle,
density_file = density.df,
chromosomes = c("chr1","chr2","chr3"),
facet_params = list(strip_position = "left"),
zoom_region = list(chr1 = c(10^8,10^8+300000),
chr2 = c(2*10^8 + 300000,2*10^8+900000),
chr3 = c(0.1*10^8,0.1*10^8+450000)),
zoom_pos = "bottom",
zoom_relheight = 0.5)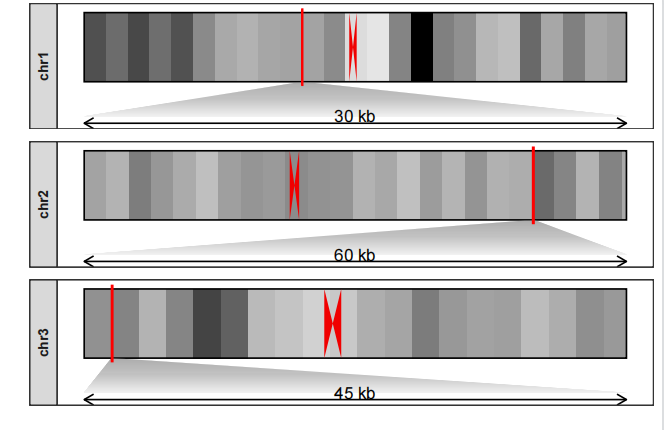
修改一下颜色:
drawChromosome(karyotype_file = karyotype.df,
centromereTri_file = centromere.triangle,
density_file = density.df,
chromosomes = c("chr1","chr2","chr3"),
facet_params = list(strip_position = "left"),
zoom_region = list(chr1 = c(10^8,10^8+300000),
chr2 = c(2*10^8 + 300000,2*10^8+900000),
chr3 = c(0.1*10^8,0.1*10^8+450000)),
zoom_pos = "bottom",
centromere_col = "#FD841F",
density_col = c("#CFF5E7","#0D4C92"),
zoom_col = c("white","#FF8DC7"))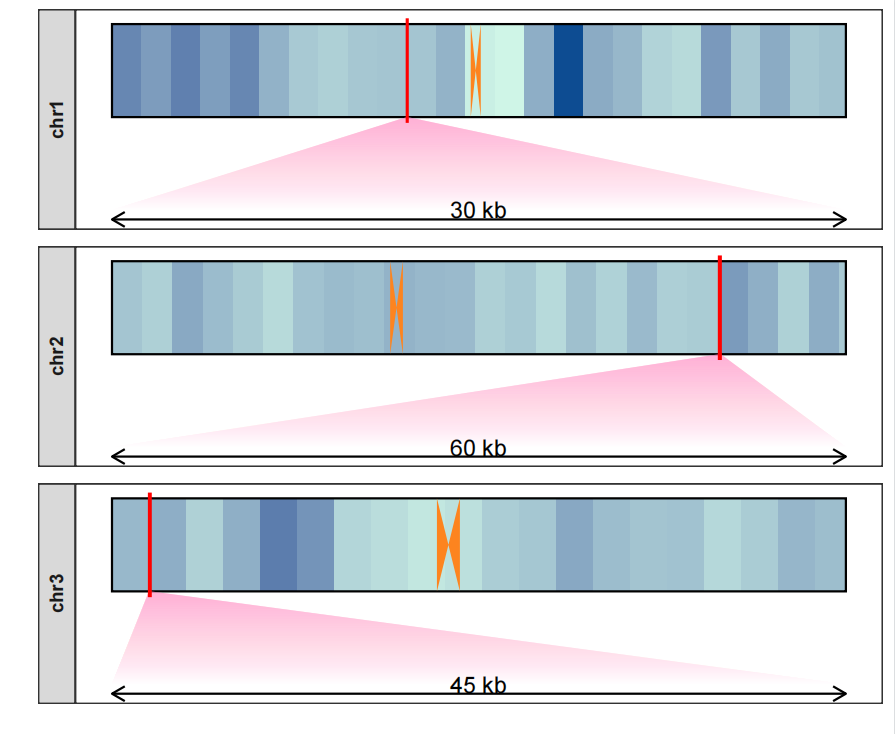
4结尾
来了就去 github 点个赞吧!
- https://github.com/junjunlab/BioSeqUtils
欢迎加入生信交流群。加我微信我也拉你进 微信群聊 老俊俊生信交流群 (微信交流群需收取 20 元入群费用,一旦交费,拒不退还!(防止骗子和便于管理)) 。QQ 群可免费加入, 记得进群按格式修改备注哦。
声明:文中观点不代表本站立场。本文传送门:https://eyangzhen.com/90687.html



